Publications
publications by categories in reversed chronological order.
2026
-
 Crystalline Dion-Jacobson 2D Layered Sn-Based Perovskites for Field-Effect TransistorsZhitian Ling , Shuanglong Wang , Arup Sarkar, Chongyao Li , Lei Gao , Ruyan Zhao , and 9 more authorsJ. Am. Chem. Soc., Mar 2026
Crystalline Dion-Jacobson 2D Layered Sn-Based Perovskites for Field-Effect TransistorsZhitian Ling , Shuanglong Wang , Arup Sarkar, Chongyao Li , Lei Gao , Ruyan Zhao , and 9 more authorsJ. Am. Chem. Soc., Mar 2026Dion-Jacobson (DJ) phase perovskites are increasingly recognized as potential semiconductor materials for electronic applications. However, the restricted coordination between the organic diammonium cations and the inorganic framework inhibits their self-assembly into efficient DJ Sn-based perovskites during solution processing. This study introduces solvent vapor assisted drop casting (SVAD) as a novel processing method to favor the organization into highly crystalline DJ perovskite films. The key aspect of the processing is the extended crystallization time in solvent vapor at elevated temperature. The processing study was conducted on four DJ perovskites that varied in the rigidity and symmetry of their A-cations to demonstrate the universal applicability of SVAD for this category of semiconductors and to establish correlations between structure and properties. The SVAD films exhibit markedly improved crystallinity and morphology compared to spin-coated samples resulting in significantly increased local and device charge carrier mobilities. DJ perovskites with rigid and symmetric A-cations form a planar [SnI6]4– octahedral framework that favors structural order and charge carrier transport. This work paves the way for the solution processing of highly ordered DJ Sn-based perovskites for their implementation in electronic applications.
-
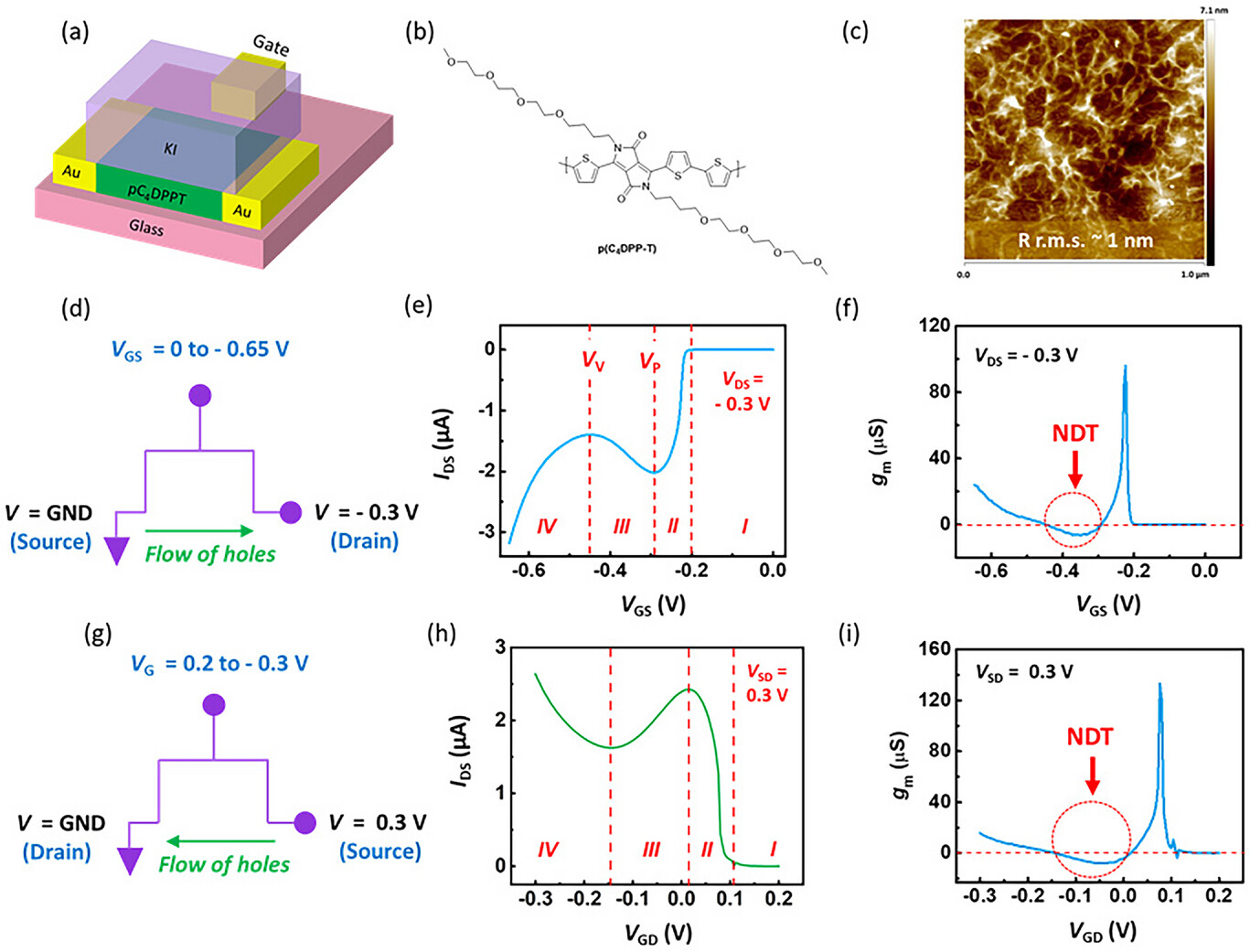 Ion-Reconfigurable “N”-Shaped Antiambipolar Behavior in Organic Electrochemical TransistorsDebdatta Panigrahi , Daniele Zucchelli , Zeinab Hamid , Christina Kousseff , Arup Sarkar, Aristea Pavlou , and 6 more authorsAdvanced Materials, Jan 2026
Ion-Reconfigurable “N”-Shaped Antiambipolar Behavior in Organic Electrochemical TransistorsDebdatta Panigrahi , Daniele Zucchelli , Zeinab Hamid , Christina Kousseff , Arup Sarkar, Aristea Pavlou , and 6 more authorsAdvanced Materials, Jan 2026Abstract “N”-shaped negative differential transconductance (NDT) is crucial for advanced electronic applications, including neuromorphic circuits, multivalued logic, and Logic-in-Memory devices. Organic electrochemical transistors (OECTs) offer an energy-efficient platform for these technologies, yet achieving NDT behavior in single-material OECTs remains challenging. In this study, the realization of “N”-shaped transfer characteristics in OECTs is demonstrated by combining the p(C4DPP-T) polymer with a potassium iodide (KI) electrolyte. The emergence of this unique behavior is attributed to the distinctive electrochemical properties of iodide, including its ability to participate in redox reactions and form triiodide species. Notably, this behavior emerges exclusively with iodide ions, while chloride, bromide and several other anions induce conventional monotonic p-type characteristics. By systematically tuning polymer microstructure, electrolyte concentration, and gate voltage scan rates, the intricate interplay among polymer-iodide interactions, and charge transport is uncovered, optimizing critical performance parameters such as the peak-to-valley ratio and the NDT range. Additionally, how iodine (I−) concentration effectively facilitates ion-driven reconfigurability in OECT-based circuits, enabling transitions between binary and ternary logic states is illustrated. The results highlight an unprecedented tunability of NDT behavior in OECTs and hold immense promise for the advancement of NDT-OECTs in next-generation electronic and neuromorphic applications.
-
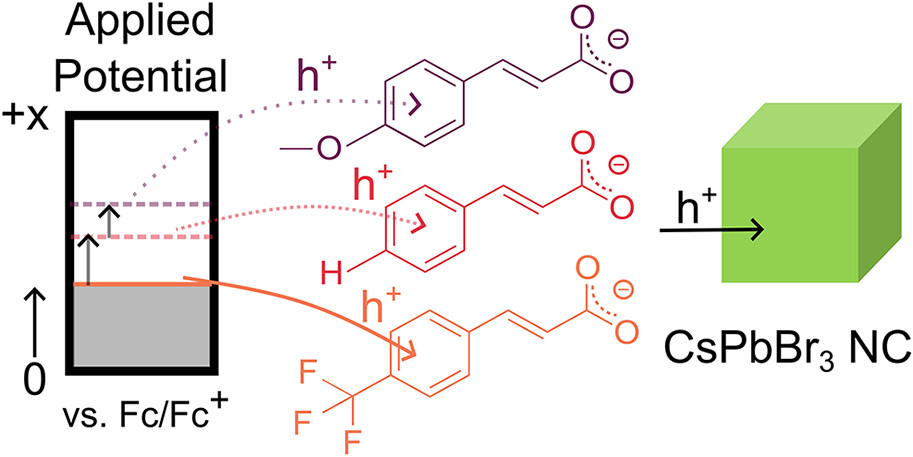 Impact of Ligand-Mediated Inductive Effects on Electrochemical p-Doping of CsPbBr3 NanocrystalsTheresa Hettiger , Roshini Jayabalan , Arup Sarkar, Jonas L. Hiller , Max Nusshör , Denis Andrienko , and 2 more authorsACS Energy Letters, Jan 2026
Impact of Ligand-Mediated Inductive Effects on Electrochemical p-Doping of CsPbBr3 NanocrystalsTheresa Hettiger , Roshini Jayabalan , Arup Sarkar, Jonas L. Hiller , Max Nusshör , Denis Andrienko , and 2 more authorsACS Energy Letters, Jan 2026Lead halide perovskite nanocrystals (NCs) are promising materials for light-emitting diodes (LEDs) due to their wavelength tunability, narrow emission line width, and high photoluminescence quantum yield. Oftentimes, these devices suffer from charge carrier imbalance and reduced charge injection because as-synthesized NCs are covered by long aliphatic ligands. Here, we report ligand exchange to small electron-withdrawing or -donating cinnamate ligands. We probe the influences of the ligands’ inductive effect on hole injection by photoluminescence spectroelectrochemistry (PL SEC). We find that hole injection into NCs covered by electron-withdrawing ligands is facilitated, and hole-only devices exhibit higher currents compared to electron donating ligands. Our work highlights the potential of PL SEC as a powerful tool to rationalize the performance of lead halide perovskite NCs in LEDs.



2025
-
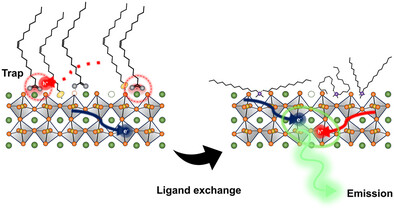 Optimizing Carrier Balance in CsPbBr3 Nanocrystal LEDs: The Role of Alkyl Ligands and Polar Electron Transport LayersRoshini Jayabalan , Girish K. Hanumantharaju , Theresa Hettiger , Arup Sarkar, Fengshuo Zu , Aladin Ullrich , and 6 more authorsAdvanced Optical Materials, Aug 2025
Optimizing Carrier Balance in CsPbBr3 Nanocrystal LEDs: The Role of Alkyl Ligands and Polar Electron Transport LayersRoshini Jayabalan , Girish K. Hanumantharaju , Theresa Hettiger , Arup Sarkar, Fengshuo Zu , Aladin Ullrich , and 6 more authorsAdvanced Optical Materials, Aug 2025Abstract The study of lead halide perovskite nanocrystal based light-emitting diodes (LEDs) has advanced significantly, with notable improvements in stability and optical properties. However, optimizing charge carrier injection and transport remains a challenge. Efficient electroluminescence requires a balanced transport of both holes and electrons within the emitting material. Here, cubic CsPbBr3 nanocrystals passivated with oleylamine and oleic acid are investigated, comparing them to ligand-exchanged nanocrystals with didodecyldimethylammonium bromide (DDABr). Nuclear magnetic resonance spectroscopy and transmission electron microscopy confirm successful ligand exchange, revealing reduced ligand coverage in DDABr-treated nanocrystals. Photoelectron spectroscopy, spectroelectrochemistry, and single-carrier devices indicate improved hole injection in DDABr-capped nanocrystals. Density functional theory calculations further reveal the influence of ligand type and coverage on energy levels, with oleic acid introducing localized states in native nanocrystals. Additionally, incorporation of a polar electron transport layer enhances LED performance by over an order of magnitude in DDABr-capped nanocrystals, driven by improved charge balance arising from the spontaneous orientation polarization of the electron transport layer. These findings highlight the critical role of ligand selection, passivation degree, and charge transport control by the adjacent organic transport layers in optimizing LED efficiency.
-
 A Multireference Picture of Electronic Excited States in Vanadyl and Copper Tetraphenyl Porphyrin Molecular QubitsArup Sarkar, and Alessandro LunghiThe Journal of Physical Chemistry A, Jul 2025
A Multireference Picture of Electronic Excited States in Vanadyl and Copper Tetraphenyl Porphyrin Molecular QubitsArup Sarkar, and Alessandro LunghiThe Journal of Physical Chemistry A, Jul 2025The nature of electronic excited states has a deep impact on the dynamics of molecular spins but remains poorly understood and characterized. Here we carry out a thorough multiconfigurational investigation for two prototypical molecular qubits based on vanadyl and copper tetraphenyl porphyrins. State-average CASSCF and NEVPT2 calculations have been employed with four different active spaces of growing complexity to account for the d–d, second d-shell, ligand-to-metal charge transfer states, and π–π* excited states, revealing an in-depth picture of low-lying excited states in agreement with experimental observations. The largest active spaces attempted (13,14) for the vanadyl and (17,12) for the copper compounds reveal that the lowest-lying excited states originate from π–π* quartet excitations. These findings shed light on the nature of the excited states of molecular qubits, taking an important step toward elucidating their role in molecular spin dynamics as well as determining novel strategies for the optical read-out of spin states.
-
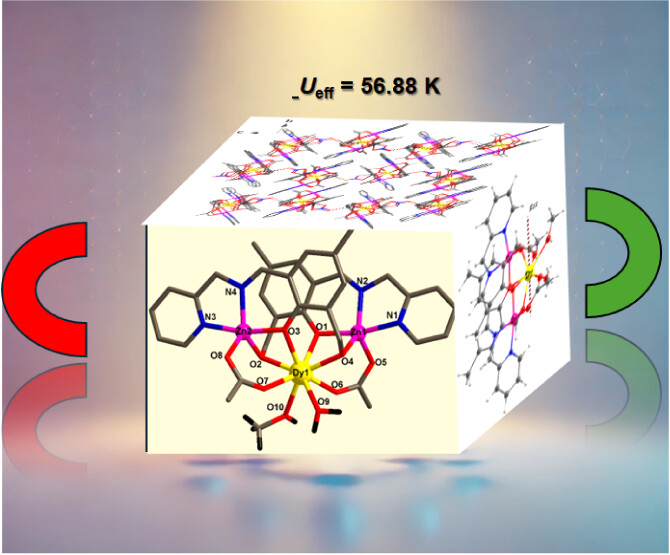 Trinuclear ZnII-LnIII Complexes: Single-Ion Magnet Behavior in the Zn2Dy AnalogueSoumalya Roy , Botan Li , Ezhava Manu Manohar , Arup Sarkar, Sukanta Saha , Lin Sun , and 3 more authorsCrystal Growth & Design, Mar 2025
Trinuclear ZnII-LnIII Complexes: Single-Ion Magnet Behavior in the Zn2Dy AnalogueSoumalya Roy , Botan Li , Ezhava Manu Manohar , Arup Sarkar, Sukanta Saha , Lin Sun , and 3 more authorsCrystal Growth & Design, Mar 2025Placing diamagnetic transition metal ions in the vicinity of lanthanide ions separated by a single coordinated oxygen atom has emerged as an effective strategy to suppress quantum tunneling of magnetization (QTM), thereby enhancing the performance of single-molecule magnets. In this context, we have synthesized trinuclear ZnII-LnIII [LnZn2L2(CH3COO)2(H2O)(CH3OH)]·NO3 (Ln= Dy(1) and Gd(2)) complexes employing a multisite coordinating Schiff base ligand, (E)-2-(hydroxymethyl)-4-methyl-6-[(pyridine-2-ylmethyl)iminomethyl]phenol, in the presence of Ln(NO3)3.xH2O and Zn(OAc)2·2H2O. The complexes crystallize in the monoclinic crystal system with the P21/c space group. The trimetallic system mainly consists of two Zn ions, one Ln ion, two deprotonated ligands, a solvent molecule (CH3OH), aqua ligands, and NO3– present outside the coordination sphere. Magnetic studies demonstrate complex 1 as a single ion magnet with Ueff = 56.88 K and pre-exponential parameter τ = 1.94 × 10–6 s. State-average CASSCF/RASSI-SO calculations performed on the Zn2Dy complex reveal a large magnetization reversal barrier of 375 cm–1 which is in accordance with the field-induced SMM behavior of this complex.



2024
-
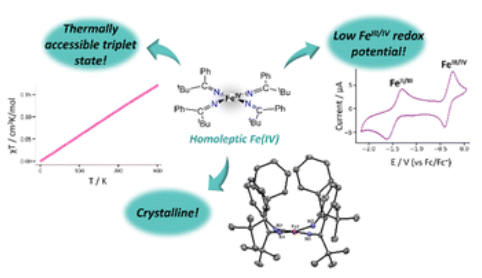 A Homoleptic Fe(IV) Ketimide Complex with a Low-Lying Excited StatePhoebe R. Hertler , Arturo Vega , Andrea Darù , Arup Sarkar, Richard A. Lewis , Guang Wu , and 2 more authorsChem. Sci., Sep 2024
A Homoleptic Fe(IV) Ketimide Complex with a Low-Lying Excited StatePhoebe R. Hertler , Arturo Vega , Andrea Darù , Arup Sarkar, Richard A. Lewis , Guang Wu , and 2 more authorsChem. Sci., Sep 2024Reaction of 4 equiv of Li(N=C(tBu)Ph) with FeIICl2 results in isolation of [Li(Et2O)]2[FeII(N=C(tBu)Ph)4] (1), in good yields. Reaction of 1 with 1 equiv of I2 leads to formation of [FeIV(N=C(tBu)Ph)4] (2), in moderate yields. 57Fe Mössbauer spectroscopy confirms the Fe(IV) oxidation state of 2, and X-ray crystallography reveals that 2 has a square planar coordination geometry along with several intramolecular H···C interactions. Furthermore, SQUID magnetometry indicates a small magnetic moment at room temperature, suggestive of accessible S = 1 state. Both density functional theory and multiconfigurational calculations were done to elucidate the nature of the ground state. Consistent with the experimental results, the ground state was found to be a closed-shell S = 0 state with an S = 1 excited state close in energy.

2023
-
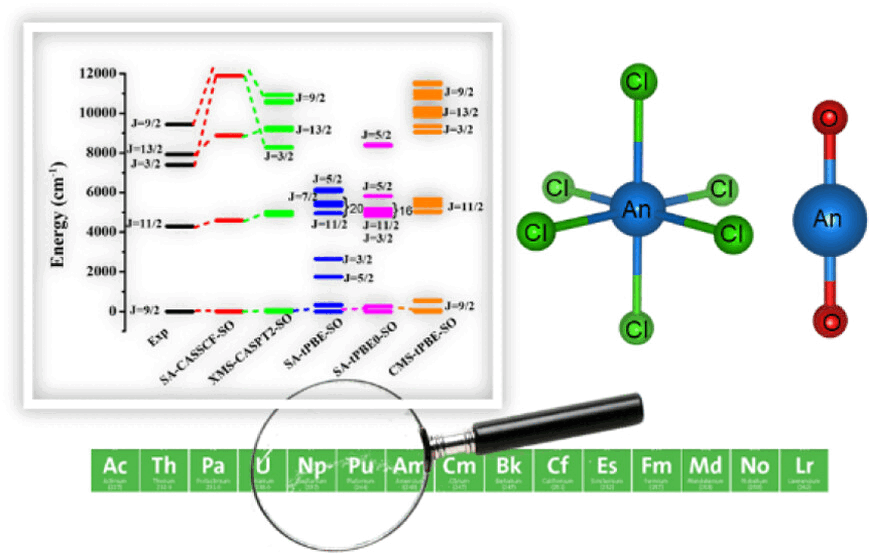 Multiconfiguration Pair-Density Functional Theory for Vertical Excitation Energies in Actinide MoleculesArup Sarkar, and Laura GagliardiThe Journal of Physical Chemistry A, Oct 2023
Multiconfiguration Pair-Density Functional Theory for Vertical Excitation Energies in Actinide MoleculesArup Sarkar, and Laura GagliardiThe Journal of Physical Chemistry A, Oct 2023Modeling actinides with electronic structure theories is challenging because these systems present a strong ligand field and metal-ligand covalency. We systematically investigate the effectiveness of pair-density functional theory (PDFT) for the calculation of vertical excitation energies in An(III), [AnIIICl6]3-, and [AnVIO2]2+ (An = U, Np, Pu, and Am). We compare the performance of PDFT, hybrid PDFT, and multistate PDFT with traditional active-space methods followed by perturbation theory, like multistate CASPT2, and with experimental data. Overall, multistate PDFT gives quantitative agreement with multistate CASPT2 at a significantly reduced computational cost.
-
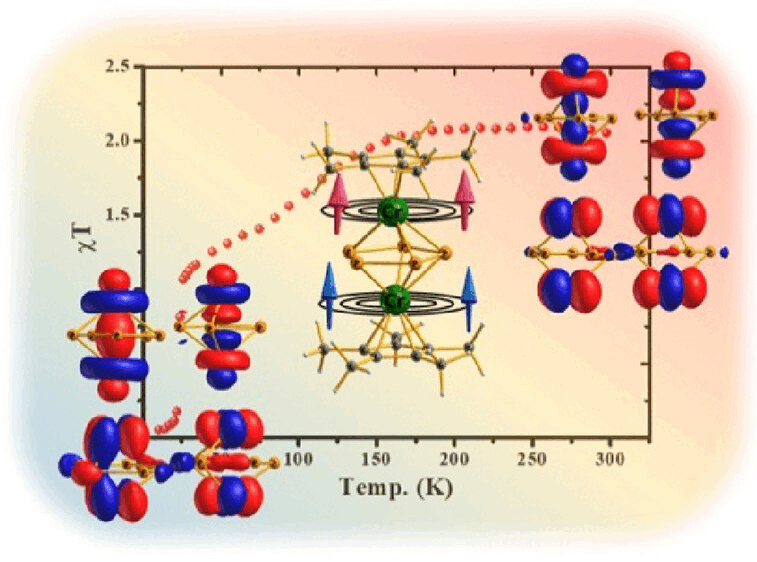 Understanding Antiferromagnetic and Ligand Field Effects on Spin Crossover in a Triple-Decker Dimeric Cr(II) ComplexArup Sarkar, Matthew R. Hermes , Christopher J. Cramer , John S. Anderson , and Laura GagliardiJournal of the American Chemical Society, Oct 2023
Understanding Antiferromagnetic and Ligand Field Effects on Spin Crossover in a Triple-Decker Dimeric Cr(II) ComplexArup Sarkar, Matthew R. Hermes , Christopher J. Cramer , John S. Anderson , and Laura GagliardiJournal of the American Chemical Society, Oct 2023Two possible explanations for the temperature dependence of spin-crossover (SCO) behavior in the dimeric triple-decker Cr(II) complex ([(\eta5-C5Me5)Cr(\mu2:\eta5-P5)Cr(\eta5-C5Me5)]+) have been offered. One invokes variations in antiferromagnetic interactions between the two Cr(II) ions, whereas the other posits the development of a strong ligand-field effect favoring the low-spin ground state. We perform multireference electronic structure calculations based on the multiconfiguration pair-density functional theory to resolve these effects. We find quintet, triplet, and singlet electronic ground states, respectively, for the experimental geometries at high, intermediate, and low temperatures. The ground-state transition from quintet to triplet at an intermediate temperature derives from increased antiferromagnetic interactions between the two Cr(II) ions. By contrast, the ground-state transition from triplet to singlet at low temperature can be attributed to increased ligand-field effects, which dominate with continued variations in antiferromagnetic coupling. This study provides quantitative detail for the degree to which these two effects can act in concert for the observed SCO behavior in this complex and others subject to temperature-dependent variations in geometry.
-
 Ab Initio Modelling of Lanthanide-Based Molecular Magnets: Where to from Here?Sourav Dey , Tanu Sharma , Arup Sarkar, and Gopalan RajaramanAug 2023
Ab Initio Modelling of Lanthanide-Based Molecular Magnets: Where to from Here?Sourav Dey , Tanu Sharma , Arup Sarkar, and Gopalan RajaramanAug 2023Ab initio calculations have played an active role in the design and development of Lanthanide-based single-ion magnets (SIMs) for the last two decades or so. These methods not only offer insight into the molecules that are reported but also hold a significant predictive potential to take this area forward. In this chapter, we aim to give an overview of the electronic structureElectronic structure method (ab initio SA-CASSCFComplete Active Space Self-Consistent Field (CASSCF)/RASSI-SO/SINGLE_ANISOSingle_ANISO) to interpret, analyse and predict the magnetic properties of lanthanide-based SMMs. In the past few years, we have witnessed the evolution of blocking temperature (TB) and blocking barrierBlocking barrier (Ueff) of Dysprosium-based Lanthanide SIMs. Among other classes of molecules that have intriguing magnetic properties, a class of molecules with a higher-order Dnh symmetry (n\thinspace=\thinspace4, 5, 6 and 8) were predicted to yield superior magnetic properties and were later synthesised and found to possess very large Ueff values and decent blocking temperatures. Particularly, molecules with D5h symmetry played an important role in understanding various contributions to the barrier height, and more than fifty molecules are reported now, with several variations yielding the absence of SIM behaviour to some of the best. In this chapter, we aim to give an overview of the variation observed in this class and another new class of molecules, i.e. Ln-encapsulated fullerene as SMMs. Particularly, we aim to showcase the strength of these methods in understanding the mechanism of magnetisation relaxation and the role of symmetry in dictating the desired magnetic characteristics.
-
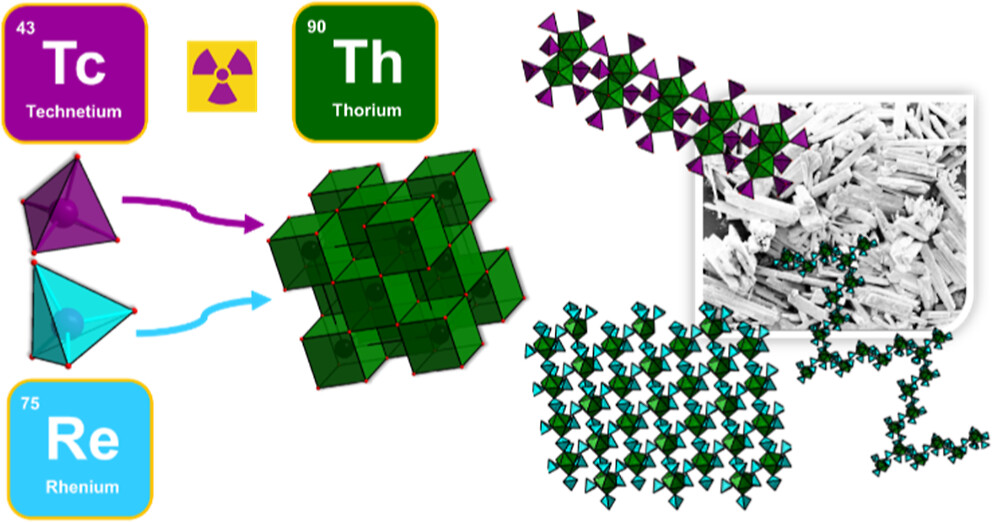 Elucidating Actinide-Pertechnetate and Actinide-Perrhenate Bonding via a Family of Th-TcO4 and Th-ReO4 Frameworks and SolutionsMohammad Shohel , Jenna Bustos , Gautam D. Stroscio , Arup Sarkar, and May NymanInorganic Chemistry, Aug 2023
Elucidating Actinide-Pertechnetate and Actinide-Perrhenate Bonding via a Family of Th-TcO4 and Th-ReO4 Frameworks and SolutionsMohammad Shohel , Jenna Bustos , Gautam D. Stroscio , Arup Sarkar, and May NymanInorganic Chemistry, Aug 2023Technetium-99, a β-emitter produced from 235U fission, poses a challenge for the nuclear industry due to co-extraction of pertechnetate (TcO4-) with the actinides (An) during nuclear fuel reprocessing. Previous studies suggested that direct coordination of pertechnetate with An plays an important role in the coextraction process. However, few studies have provided direct evidence for An-TcO4- bonding in the solid state, and even fewer in solution. The present study describes synthesis and structural elucidation of a family of thorium(IV)-pertechnetate/perrhenate (ReO4-, nonradioactive surrogate) compounds, which is obtained by dissolution of thorium oxyhydroxide in perrhenic/pertechnic acid followed by crystallization, with or without heating. For reaction ratios of 3:1, 4:1, and 6:1 MO4-/Th(IV) (M = Tc, Re), the crystallized compounds reflect the same ratio, suggesting facile and flexible coordination. Nine structures reveal 1-dimensional and 2-dimensional frameworks with varying topologies. While a multitude of compounds isolated from 4:1 (and 6:1) reaction solutions feature Th monomers linked by MO4-, the 3:1 reaction solution yielded the well-known dihydroxide-bridged thorium dimer, linked, and capped by MO4-. Density functional theory calculations on ReO4-/TcO4- isomorphs suggest similar bonding characteristics in the solid state, but experimental solution characterization noted differences. Specifically, small-angle X-ray scattering studies suggest the bonding of Th-TcO4- persists in solution, while Th-ReO4- bonding is less apparent.
-
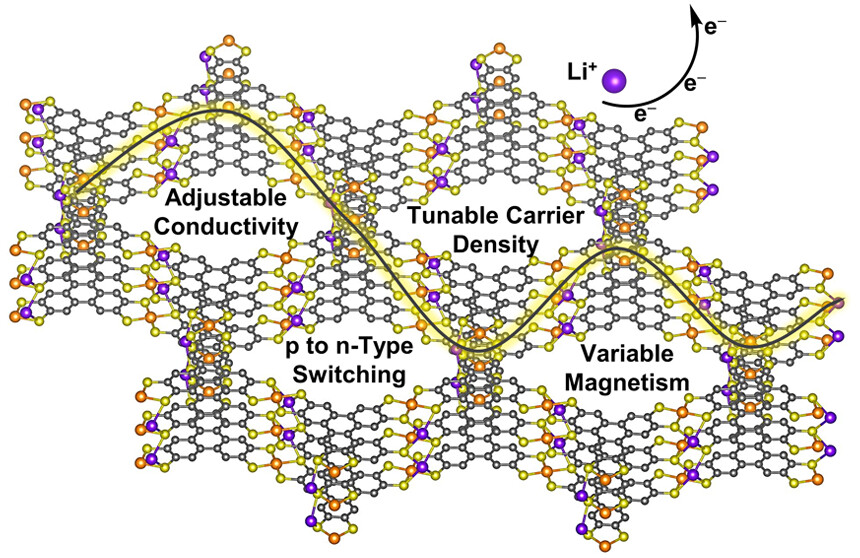 Broad Electronic Modulation of Two-Dimensional Metal–Organic Frameworks over Four Distinct Redox StatesLei Wang , Arup Sarkar, Garrett L Grocke , Daniel William Laorenza , Baorui Cheng , Andrew Ritchhart , and 4 more authorsJournal of the American Chemical Society, Apr 2023
Broad Electronic Modulation of Two-Dimensional Metal–Organic Frameworks over Four Distinct Redox StatesLei Wang , Arup Sarkar, Garrett L Grocke , Daniel William Laorenza , Baorui Cheng , Andrew Ritchhart , and 4 more authorsJournal of the American Chemical Society, Apr 2023Two-dimensional (2D) inorganic materials have emerged as exciting platforms for (opto)electronic, thermoelectric, magnetic, and energy storage applications. However, electronic redox tuning of these materials can be difficult. Instead, 2D metal–organic frameworks (MOFs) offer the possibility of electronic tuning through stoichiometric redox changes, with several examples featuring one to two redox events per formula unit. Here, we demonstrate that this principle can be extended over a far greater span with the isolation of four discrete redox states in the 2D MOFs LixFe3(THT)2 (x = 0–3, THT = triphenylenehexathiol). This redox modulation results in 10,000-fold greater conductivity, p- to n-type carrier switching, and modulation of antiferromagnetic coupling. Physical characterization suggests that changes in carrier density drive these trends with relatively constant charge transport activation energies and mobilities. This series illustrates that 2D MOFs are uniquely redox flexible, making them an ideal materials platform for tunable and switchable applications.
-
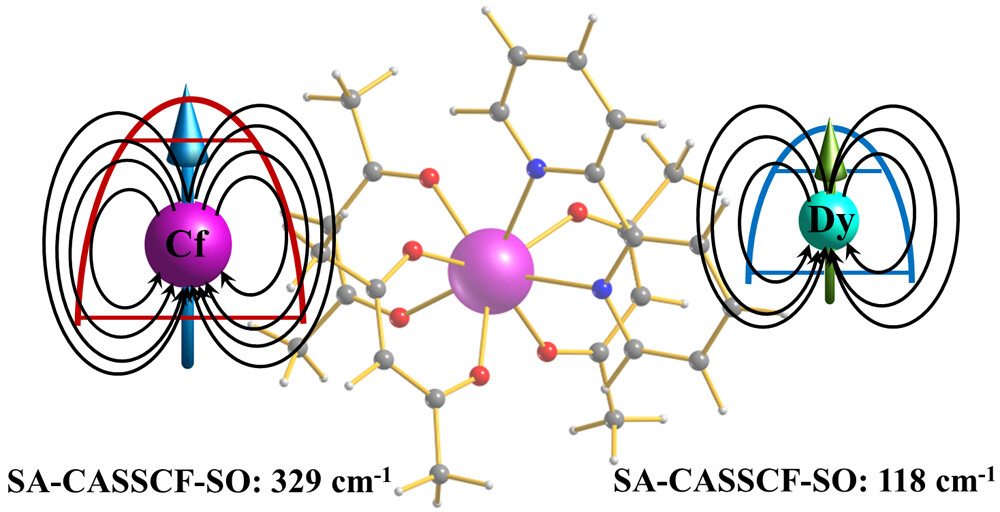 Theoretical Investigation of Single-Molecule-Magnet Behavior in Mononuclear Dysprosium and Californium ComplexesDebmalya Ray , Meagan S. Oakley , Arup Sarkar, Xiaojing Bai , and Laura GagliardiInorganic Chemistry, Apr 2023
Theoretical Investigation of Single-Molecule-Magnet Behavior in Mononuclear Dysprosium and Californium ComplexesDebmalya Ray , Meagan S. Oakley , Arup Sarkar, Xiaojing Bai , and Laura GagliardiInorganic Chemistry, Apr 2023Early-actinide-based (U, Np, and Pu) single-molecule magnets (SMMs) have yet to show magnetic properties similar to those of highly anisotropic lanthanide-based ones. However, there are not many studies exploring the late-actinides (more than half-filled f shells) as potential candidates for SMM applications. We computationally explored the electronic structure and magnetic properties of a hypothetical Cf(III) complex isostructural to the experimentally synthesized Dy(dbm)3(bpy) complex (bpy = 2,2′-bipyridine; dbm = dibenzoylmethanoate) via multireference methods and compared them to those of the Dy(III) analogue. This study shows that the Cf(III) complex can behave as a SMM and has a greater magnetic susceptibility compared to other experimentally and computationally studied early-actinide-based (U, Np, and Pu) magnetic complexes. However, Cf spontaneously undergoes α-decay and converts to Cm. Thus, we also explored the isostructural Cm(III)-based complex. The computed magnetic susceptibility and g-tensor values show that the Cm(III) complex has poor SMM behavior in comparison to both the Dy(III) and Cf(III) complexes, suggesting that the performance of Cf(III)-based magnets may be affected by α-decay and can explain the poor performance of experimentally studied Cf(III)-based molecular magnets in the literature. Further, this study suggests that the ligand field is dominant in Cf(III), which helps to increase the magnetization blocking barrier by nearly 3 times that of its 4f congener.
-
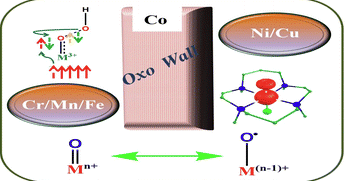 Theoretical study of the formation of metal–oxo species of the first transition series with the ligand 14-TMC: driving factors of the “Oxo Wall”Monika , Manjeet Kumar , Somi , Arup Sarkar, Manoj Kumar Gupta , and Azaj AnsariDalton Trans., Sep 2023
Theoretical study of the formation of metal–oxo species of the first transition series with the ligand 14-TMC: driving factors of the “Oxo Wall”Monika , Manjeet Kumar , Somi , Arup Sarkar, Manoj Kumar Gupta , and Azaj AnsariDalton Trans., Sep 2023Terminal metal–oxo species of the early transition metal series are well known, whereas those for the late transition series are rare, and this is related to the “Oxo Wall”. Here, we have undertaken a theoretical study on the formation of metal–oxo species from the metal hydroperoxo species of the 3d series (Cr, Mn, Fe, Co, Ni, and Cu) with the ligand 14-TMC (1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane) via O⋯O bond cleavage. DFT calculations reveal that the barrier for O⋯O bond cleavage is higher with the late transition metals (Co, Ni, and Cu) than the early transition metals (Cr, Mn, and Fe), and the formed late metal–oxo species are also thermodynamically less stable. The higher barrier may be due to electronic repulsion because of the pairing of d electrons. In the late transition metal series, the electron goes into an antibonding orbital, which decreases the bond order and hence decreases the possibility of metal–oxo formation. Computed structural parameters and spin densities suggest that valence tautomerism occurs in the late transition metal–oxo species which remain as a metal–oxyl. Our findings support the concept of the “Oxo Wall”.







2022
-
 Implementing the Ambiphilicity of an Organotellurenyl Cation for the Synthesis of a Platinum(II)-Based Carboxylate-Bridged Heterobimetallic Complex: Structure and Bonding AnalysisRajesh Deka , Arup Sarkar, Ray J. Butcher , and Harkesh B. SinghEuropean Journal of Inorganic Chemistry, Aug 2022
Implementing the Ambiphilicity of an Organotellurenyl Cation for the Synthesis of a Platinum(II)-Based Carboxylate-Bridged Heterobimetallic Complex: Structure and Bonding AnalysisRajesh Deka , Arup Sarkar, Ray J. Butcher , and Harkesh B. SinghEuropean Journal of Inorganic Chemistry, Aug 2022Abstract The metathesis reaction of aryltellurium chloride, ppyTeCl [ppy=2-(2’-pyridyl)phenyl] (5) with AgClO4 afforded Intramolecular Chalcogen Bonding (IChB)-stabilized organotellurenyl cation [ppyTe] ⋅ ClO4 ([6] ⋅ ClO4). The reaction of [6] ⋅ ClO4 with K2PtCl4 in acetic acid resulted in the formation of platinum(II)-based carboxylate-bridged heterobimetallic complex [Pt(ppyTe)(μ-OOCMe)Cl2], 7 containing an organotellurenyl cation as ligand. This is the first time a Pt(II) complex of an organotellurenyl cation donor-acceptor ligand has been reported. Complex 7 was characterized by multi-nuclear NMR spectroscopy (1H, 13C, and 125Te), FT-IR spectroscopy, CHN analysis, and single-crystal X-ray diffraction studies. The single-crystal X-ray diffraction studies and DFT analysis infer that the Te center in 7 behaves as an ambiphilic ligand, accepting electrons from the N-atom of the ppy moiety and the O-atom of the carboxylate groups while simultaneously donating electrons to the Pt(II) center. The 2D-fingerprint plots derived from the Hirshfeld surface analysis reveal multiple intermolecular interactions in the packing diagrams of complex 7. The natural charges accumulated on the atoms in complex 7 were calculated by Natural Population Analysis (NPA). In addition, the HOMO-LUMO energy gap in 7 was calculated.
-
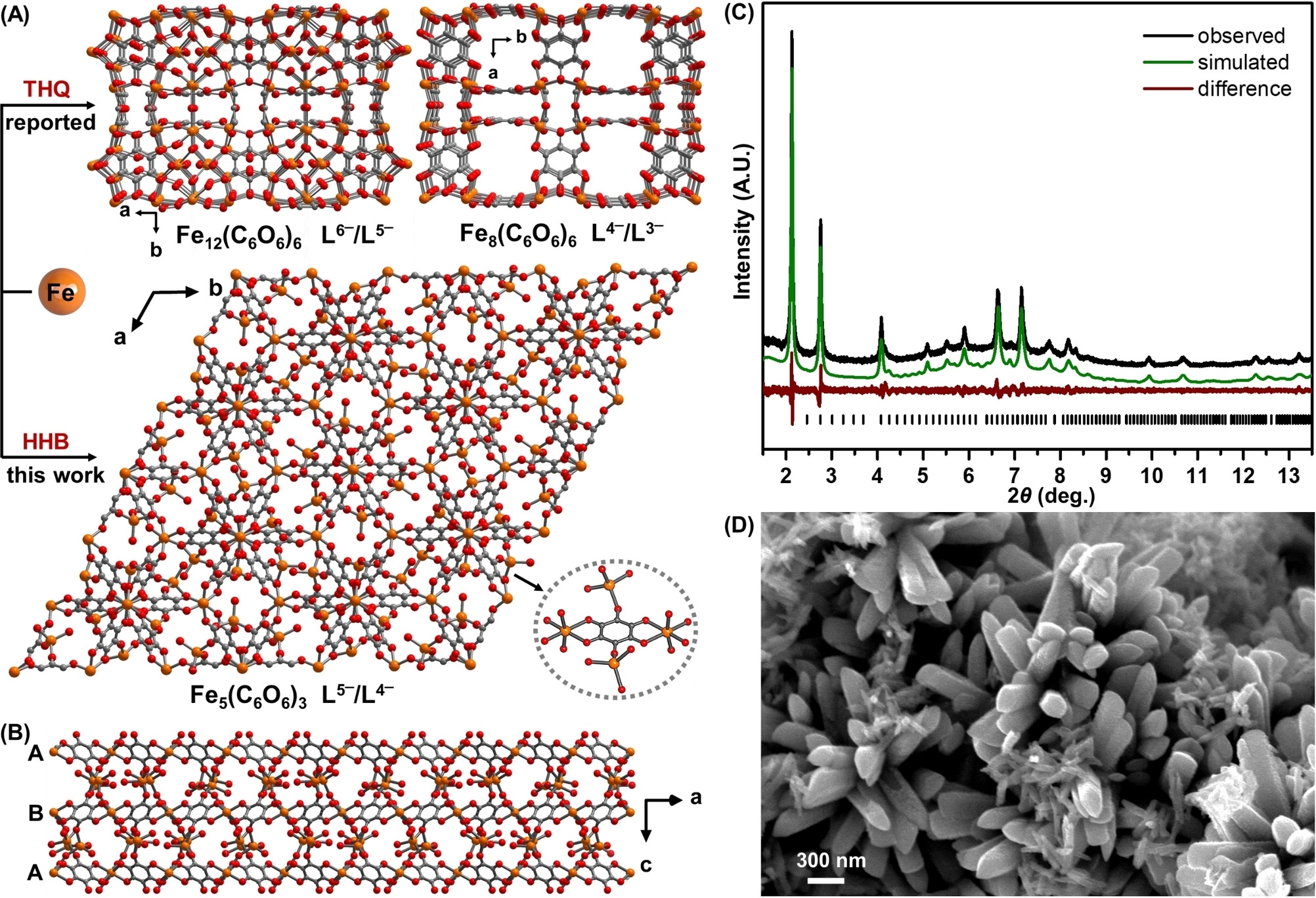 Linker Redox Mediated Control of Morphology and Properties in Semiconducting Iron-Semiquinoid Coordination Polymers**Lei Wang , Robert J Papoular , Noah E Horwitz , Jiaze Xie , Arup Sarkar, Dario Campisi , and 7 more authorsAngewandte Chemie International Edition, Sep 2022
Linker Redox Mediated Control of Morphology and Properties in Semiconducting Iron-Semiquinoid Coordination Polymers**Lei Wang , Robert J Papoular , Noah E Horwitz , Jiaze Xie , Arup Sarkar, Dario Campisi , and 7 more authorsAngewandte Chemie International Edition, Sep 2022Abstract The emergence of conductive 2D and less commonly 3D coordination polymers (CPs) and metal?organic frameworks (MOFs) promises novel applications in many fields. However, the synthetic parameters for these electronically complex materials are not thoroughly understood. Here we report a new 3D semiconducting CP Fe5(C6O6)3, which is a fusion of 2D Fe-semiquinoid materials and 3D cubic Fex(C6O6)y materials, by using a different initial redox-state of the C6O6 linker. The material displays high electrical conductivity (0.02?S?cm?1), broad electronic transitions, promising thermoelectric behavior (S2σ=7.0?10?9?W?m?1?K?2), and strong antiferromagnetic interactions at room temperature. This material illustrates how controlling the oxidation states of redox-active components in conducting CPs/MOFs can be a ?pre-synthetic? strategy to carefully tune material topologies and properties in contrast to more commonly encountered post-synthetic modifications.
-
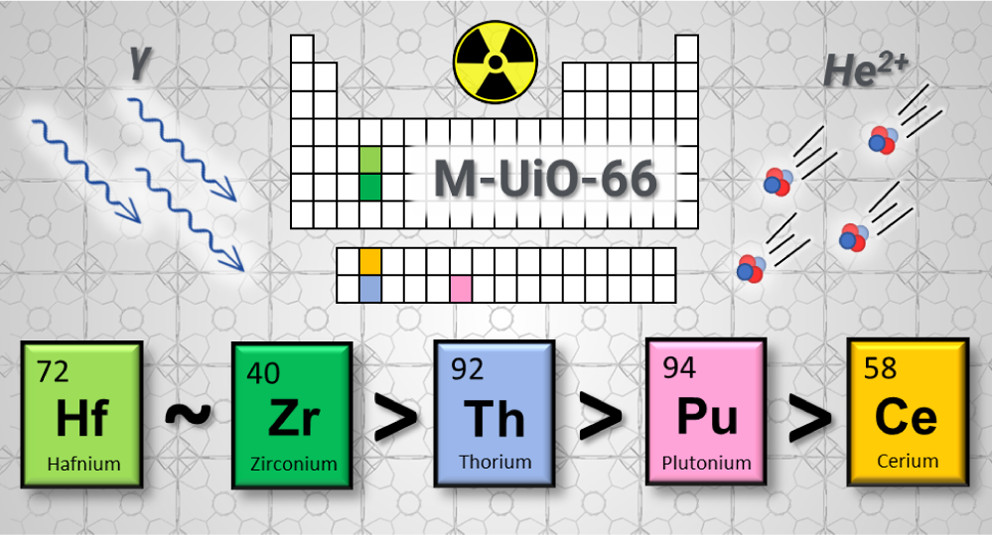 Role of Metal Selection in the Radiation Stability of Isostructural M-UiO-66 Metal-Organic FrameworksAshley M. Hastings , Melissa Fairley , Megan C. Wasson , Dario Campisi , Arup Sarkar, Zoë C. Emory , and 9 more authorsChemistry of Materials, Sep 2022
Role of Metal Selection in the Radiation Stability of Isostructural M-UiO-66 Metal-Organic FrameworksAshley M. Hastings , Melissa Fairley , Megan C. Wasson , Dario Campisi , Arup Sarkar, Zoë C. Emory , and 9 more authorsChemistry of Materials, Sep 2022Robust and versatile metal-organic frameworks (MOFs) have emerged as sophisticated scaffolds to meet the critical needs of the nuclear community, but their performance depends on their underexplored structural integrities in high-radiation fields. The contributions of selected metal nodes in the radiation stability of MOFs within the isostructural M-UiO-66 series (where M = Zr, Ce, Hf, Th, and Pu; Zr-UiO-66 experiments were executed in a previous work) have been determined. Ce-, Hf-, and Th-UiO-66 MOF samples were irradiated via gamma and He-ion methodologies to obtain doses up to 3 MGy and 85 MGy, respectively, the latter strikingly higher than that obtained in most other studies. Appreciable self-irradiation constituted the total absorbed doses, up to 31 MGy of the gamma-irradiated Pu-UiO-66 samples. Structural degradation was ascertained by powder X-ray diffraction, X-ray total scattering, vibrational spectroscopy, and, where possible, N2 physisorption isotherms. Diffuse reflectance infrared Fourier transform spectroscopy provided atomic-level mechanistic insights to reveal that the node-linker connection was most susceptible to radiation damage. Density functional theory calculations were performed on cluster models to evaluate the binding energy of the linkers to each metal node. While the isostructures disclosed the same breakdown signatures, distinct radiation sensitivity as a function of metal selection was evident and followed the trend Hf-UiO-66 ∼Zr-UiO-66 > Th-UiO-66 > Pu-UiO-66 > Ce-UiO-66. We anticipate that these endeavors will contribute to the rational design of radiation-resistant materials for targeted applications.
-
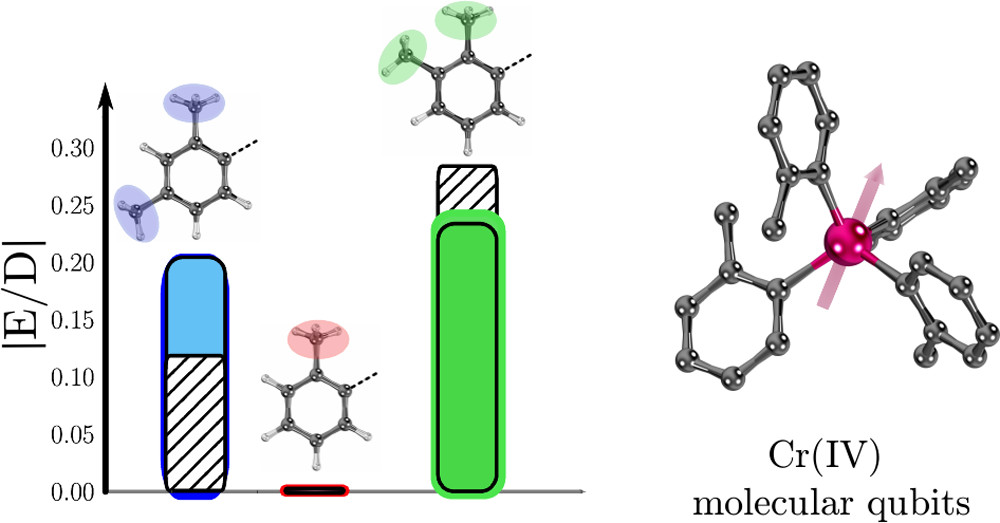 Multiconfiguration Pair-Density Functional Theory for Chromium(IV) Molecular QubitsArturo Vega , Riddhish Pandharkar , Gautam D. Stroscio , Arup Sarkar, Donald G. Truhlar , and Laura GagliardiJACS Au, Sep 2022
Multiconfiguration Pair-Density Functional Theory for Chromium(IV) Molecular QubitsArturo Vega , Riddhish Pandharkar , Gautam D. Stroscio , Arup Sarkar, Donald G. Truhlar , and Laura GagliardiJACS Au, Sep 2022Pseudotetrahedral organometallic complexes containing chromium(IV) and aryl ligands have been experimentally identified as promising molecular qubit candidates. Here we present a computational protocol based on multiconfiguration pair-density functional theory for computing singlet-triplet gaps and zero-field splitting (ZFS) parameters in Cr(IV) aryl complexes. We find that two multireference methods, multistate complete active space second-order perturbation theory (MS-CASPT2) and hybrid multistate pair-density functional theory (HMS-PDFT), perform better than Kohn-Sham density functional theory for singlet-triplet gaps. Despite the very small values of the ZFS parameters, both multireference methods performed qualitatively well. MS-CASPT2 and HMS-PDFT performed particularly well for predicting the trend in the ratio of the rhombic and axial ZFS parameters, |E/D|. We have also investigated the dependence and sensitivity of the calculated ZFS parameters on the active space and the molecular geometry. The methodologies outlined here can guide future prediction of ZFS parameters in molecular qubit candidates.
-
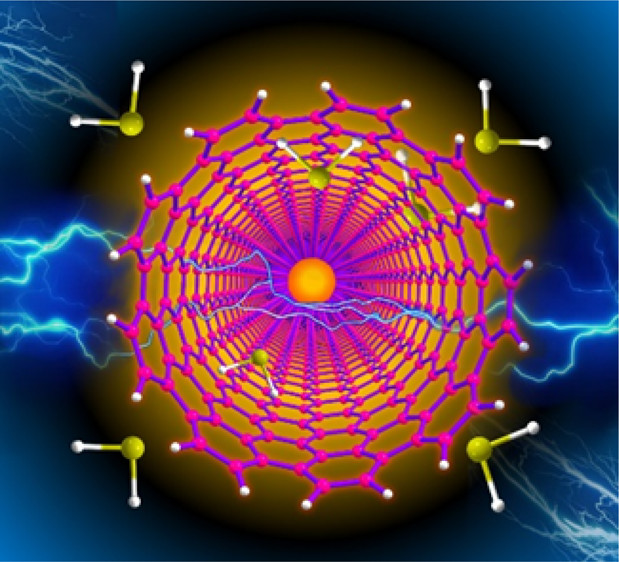 Electric-Field-Induced Solid-Gas Interfacial Chemical Reaction in Carbon Nanotube Ensembles: Route toward Ultra-sensitive Gas DetectorsItisha Dwivedi , Arup Sarkar, Gopalan Rajaraman , and Chandramouli SubramaniamACS Applied Materials and Interfaces, Mar 2022
Electric-Field-Induced Solid-Gas Interfacial Chemical Reaction in Carbon Nanotube Ensembles: Route toward Ultra-sensitive Gas DetectorsItisha Dwivedi , Arup Sarkar, Gopalan Rajaraman , and Chandramouli SubramaniamACS Applied Materials and Interfaces, Mar 2022The electric field at the sharp pointed tips of single wall carbon nanotube ensembles has been utilized to kinetically accelerate hitherto unobserved chemical reactions at the heterogeneous solid-gas interfaces. The principle of ″action-of-points″ drives specific chemical reactions between the defect sites of single wall carbon nanotubes (CNTs) and ppb levels of gaseous hydrogen sulfide. This is manifested as changes in the electrical conductivity of the conductive CNT-ensemble (cCNT) and visually tracked as enthalpic modulations at the site of the reaction through infrared thermometry. Importantly, the principle has been observed for a variety of analytes such as NH3, H2O, and H2S, leading to distinctly correlatable changes in reactivity and conductivity changes. Theoretical calculations based on the density functional theory in the presence and absence of applied electric field reveal that the applied electric field activates the H2S gas molecules by charge polarization, yielding favorable energetics. These results imply the possibility of carrying out site-specific chemical modifications for nanomaterials and also provide transformative opportunities for the development of miniaturized e-nose-based gas analyzers.
-
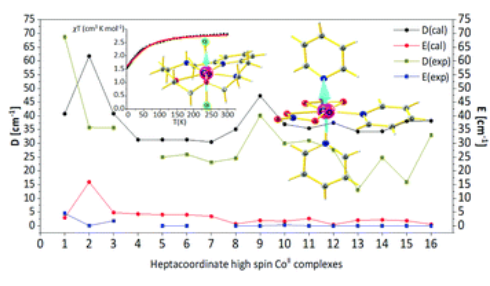 What controls the magnetic anisotropy in heptacoordinate high-spin cobalt(ii) complexes? A theoretical perspectivePeter Comba , Gopalan Rajaraman , Arup Sarkar, and Gunasekaran VelmuruganDalton Transactions, Mar 2022
What controls the magnetic anisotropy in heptacoordinate high-spin cobalt(ii) complexes? A theoretical perspectivePeter Comba , Gopalan Rajaraman , Arup Sarkar, and Gunasekaran VelmuruganDalton Transactions, Mar 2022The magnetic anisotropy of sixteen seven-coordinate high-spin CoII complexes with O, N, Cl and I donors was investigated with state-of-the-art ab initio CASSCF/NEVPT2 calculations and compared with experimental data. Based on the nature of the equatorial and axial ligands, which were found to tune the zero-field splitting, the complexes were classified into four groups. The experimental zero-field splitting parameters D which, for the various structures are in a range of +30 to +60 cm−1, as well as the g and E values are well reproduced. The investigation of the electronic structure shows that in these pentagonal bipyramidal complexes the donors and symmetry in the equatorial plane play an important role in the values of the axial zero-field splitting parameter D, and breaking of the horizontal plane of symmetry was found to enhance the magnitude of the D value. Although negative values of D are a desired condition for SIMs, many CoII based SIMs with positive zero-field splitting are fundamentally important to understand the nature of magnetic anisotropy, and seven coordinate CoII complexes with a large overall crystal field splitting might provide a way forward in this class of molecules.






2021
-
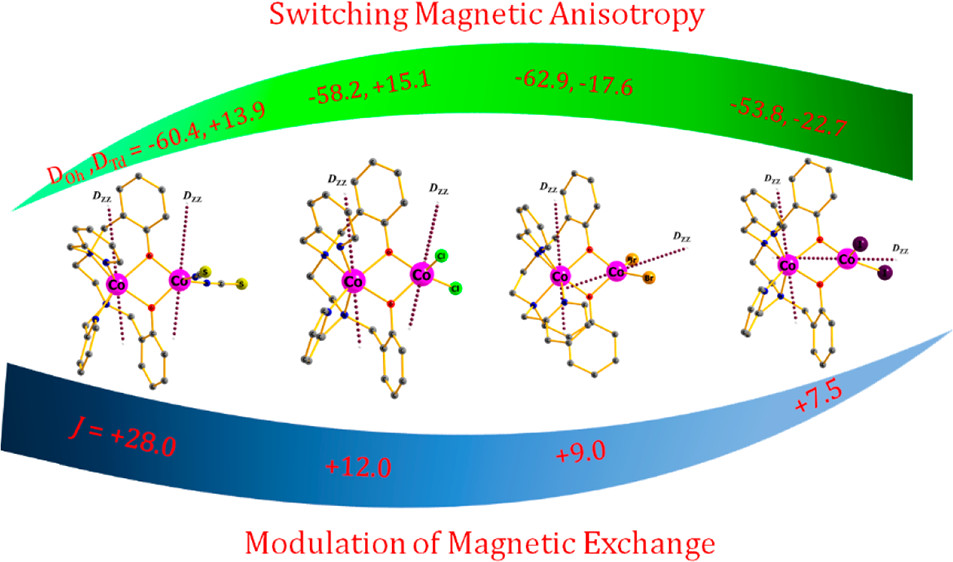 Modulation of Magnetic Anisotropy and Exchange Interaction in Phenoxide-Bridged Dinuclear Co(II) ComplexesAjit Kumar Kharwar , Arpan Mondal , Arup Sarkar, Gopalan Rajaraman , and Sanjit KonarInorganic Chemistry, Jul 2021
Modulation of Magnetic Anisotropy and Exchange Interaction in Phenoxide-Bridged Dinuclear Co(II) ComplexesAjit Kumar Kharwar , Arpan Mondal , Arup Sarkar, Gopalan Rajaraman , and Sanjit KonarInorganic Chemistry, Jul 2021We report a new class of four dimeric Co(II) complexes [Co2(bbpen)(X)2] (H2bbpen = N,N′-bis(2-hydroxybenzyl)-N,N′-bis(2-methylpyridyl)ethylenediamine) [X- = SCN (1), Cl (2), Br (3), and I (4)] with different coordination geometry of two Co(II) centers (trigonal-prismatic and pseudo-tetrahedral) and their magnetic study. Interestingly, the two Co(II) centers show two different types of magnetic anisotropy. State of the art ab initio CASSCF analysis reveals that the six-coordinate or the trigonal-prismatic Co(II) center possesses a consistently large negative axial zero-field splitting (negative D) parameter (∼-60 cm-1), while the four-coordinate or the pseudo-tetrahedral Co(II) center exhibits a range of D values from +13 to -23 cm-1. Ab initio calculations employing the lines model were used to estimate the magnetic exchange as both the Co(II) centers possess significant magnetic anisotropy. All the complexes display rare ferromagnetic interaction, and the strength of this interaction decreases as the ligand field on the pseudo-tetrahedral Co(II) center decreases from SCN- > Cl- > Br- > I-.
-
 [(VIVO)2MII5] (M = Ni, Co) Anderson wheelsHector W.L. Fraser , Emily H. Payne , Arup Sarkar, Lucinda R.B. Wilson , Dmitri Mitcov , Gary S. Nichol , and 3 more authorsDalton Transactions, Jul 2021
[(VIVO)2MII5] (M = Ni, Co) Anderson wheelsHector W.L. Fraser , Emily H. Payne , Arup Sarkar, Lucinda R.B. Wilson , Dmitri Mitcov , Gary S. Nichol , and 3 more authorsDalton Transactions, Jul 2021Heterometallic Anderson wheels of formula [(VIVO)2M II5(hmp)10Cl2](ClO4)2\textperiodcentered2MeOH (M = Ni,1; Co,2) have been synthesised from the solvothermal reaction of M(ClO4)2\textperiodcentered6H2O and VCl3with hmpH (2-(hydroxymethyl)pyridine). The metallic skeleton describes a centred hexagon, with the two vanadyl ions sitting on opposing sides of the outer ring. Magnetic susceptibility and magnetisation measurements indicate the presence of both ferromagnetic and antiferromagnetic exchange interactions. Theoretical calculations based on density functional methods reproduce both the sign and strength of the exchange interactions found experimentally, and rationalise the parameters extracted.
-
 Record High Magnetic Anisotropy in Three-Coordinate MnIII and CrII Complexes: A Theoretical PerspectiveArup Sarkar, Reshma Jose , Harshit Ghosh , and Gopalan RajaramanInorganic Chemistry, Jun 2021
Record High Magnetic Anisotropy in Three-Coordinate MnIII and CrII Complexes: A Theoretical PerspectiveArup Sarkar, Reshma Jose , Harshit Ghosh , and Gopalan RajaramanInorganic Chemistry, Jun 2021Ab initio calculations performed in two three-coordinate complexes [MnN(SiMe3)23] (1) and [K(18-crown-6) (Et2O)2][CrN(SiMe3)23] (2) reveal record-high magnetic anisotropy with the D values -64 and -15 cm-1, respectively, enlisting d4 ions back in the race for single-ion magnets. A detailed spin-vibrational analysis performed of 1 and 2 suggests dominance under barrier relaxation due to the flexible coordination spheres around the metal ion. Furthermore, several in silico models were constructed by varying the nature of donor atoms based on the X-ray structure of 1 and 2, unveiling much larger anisotropy and robust single-ion magnet (SIM) characteristics for some of the models offering design clues for low-coordinate transition-metal SIMs.
-
 Exploiting host-guest chemistry to manipulate magnetic interactions in metallosupramolecular M4L6tetrahedral cagesAaron J. Scott , Julia Vallejo , Arup Sarkar, Lucy Smythe , E. Regincós Martí , Gary S. Nichol , and 7 more authorsChemical Science, Mar 2021
Exploiting host-guest chemistry to manipulate magnetic interactions in metallosupramolecular M4L6tetrahedral cagesAaron J. Scott , Julia Vallejo , Arup Sarkar, Lucy Smythe , E. Regincós Martí , Gary S. Nichol , and 7 more authorsChemical Science, Mar 2021Reaction of Ni(OTf)2with the bisbidentate quaterpyridine ligandLresults in the self-assembly of a tetrahedral, paramagnetic cage [NiII4L6]8+. By selectively exchanging the bound triflate from [OTf⊂NiII4L6](OTf)7(1), we have been able to prepare a series of host-guest complexes that feature an encapsulated paramagnetic tetrahalometallate ion inside this paramagnetic host giving [MIIX4⊂NiII4L6](OTf)6, where MIIX42−= MnCl42−(2), CoCl42−(5), CoBr42−(6), NiCl42−(7), and CuBr42−(8) or [MIIIX4⊂NiII4L6](OTf)7, where MIIIX4−= FeCl4−(3) and FeBr4−(4). Triflate-to-tetrahalometallate exchange occurs in solution and can also be accomplished through single-crystal-to-single-crystal transformations. Host-guest complexes1-8all crystallise as homochiral racemates in monoclinic space groups, wherein the four NiN6 vertexes within a single Ni4L6unit possess the same\Deltaor\Lambdastereochemistry. Magnetic susceptibility and magnetisation data show that the magnetic exchange between metal ions in the host [NiII4] complex, and between the host and the MX4n−guest, are of comparable magnitude and antiferromagnetic in nature. Theoretically derived values for the magnetic exchange are in close agreement with experiment, revealing that large spin densities on the electronegative X-atoms of particular MX4n−guest molecules lead to stronger host-guest magnetic exchange interactions.




2020
-
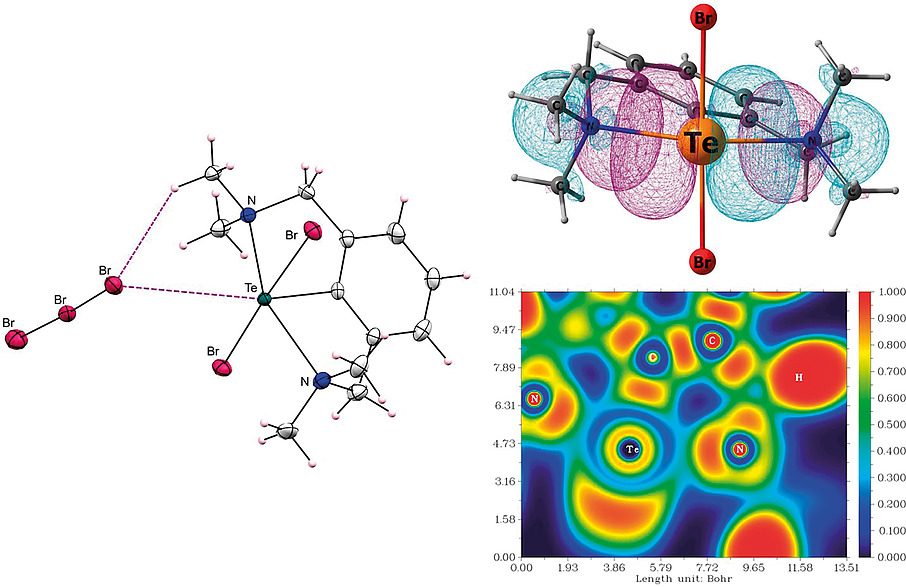 Halogenation of Diorganotelluride [2,6-(Me2NCH2)2C6H3]TenBu: Synthesis, Molecular and Electronic Structural Investigation of Monoorgano Dihalotelluronium(IV) CationRajesh Deka , Anand Gupta , Arup Sarkar, Ray J. Butcher , and Harkesh B. SinghEuropean Journal of Inorganic Chemistry, Oct 2020
Halogenation of Diorganotelluride [2,6-(Me2NCH2)2C6H3]TenBu: Synthesis, Molecular and Electronic Structural Investigation of Monoorgano Dihalotelluronium(IV) CationRajesh Deka , Anand Gupta , Arup Sarkar, Ray J. Butcher , and Harkesh B. SinghEuropean Journal of Inorganic Chemistry, Oct 2020A simple methodology for the synthesis of heteroleptic diorganotelluride [2,6-(Me2NCH2)2C6H3]TenBu (1) and homoleptic diorganotelluride [2,6-(Me2NCH2)2C6H3]2Te (2) is reported. The halogenation reactions of diorganotelluride 1 are studied. In particular, the reaction of 1 with Br2 resulted in the isolation of a monoorgano dibromotelluronium(IV) cation, namely [2,6-(Me2NCH2)2C6H3TeBr2]·Br3 ([3]·Br3). The reaction of [3]·Br3 with K2PdCl4 afforded a monoorgano mixed dihalotelluronium(IV) cation, [2,6-(Me2NCH2)2C6H3TeClBr]·PdBr4 ([4]·PdBr4). When diorganotelluride 1 was treated with I2, the reaction afforded the tellurenium(II) cation, [2,6-(Me2NCH2)2C6H3Te]·I2·I3 ([5]·I2·I3). The reaction of 1 with HgCl2 resulted in the isolation of dimeric diorganoditelluroxonium(IV) cation, [2,6-(Me2NCH2)2C6H3Te(µ-O)]2·Hg2Cl6 ([6]·Hg2Cl6). A similar diorganoditelluroxonium(IV) cation, namely [2,6-(Me2NCH2)2C6H3Te(µ-O)]2·2Br ([6]·2Br) was also obtained by the reaction of 1 with Br2 and NaOH. The crystallographic studies suggest that for each compound, both the N atoms from the pendant arms make strong Intramolecular Chalcogen Bonding (IChB) interactions with the Te center(s). The presence of a strong N→Te IChB interaction in the synthesized compounds was further validated by DFT calculations.
-
 Role of Coordination Number and Geometry in Controlling the Magnetic Anisotropy in FeII, CoII, and NiII Single-Ion MagnetsArup Sarkar, Sourav Dey , and Gopalan RajaramanChemistry - A European Journal, Jul 2020
Role of Coordination Number and Geometry in Controlling the Magnetic Anisotropy in FeII, CoII, and NiII Single-Ion MagnetsArup Sarkar, Sourav Dey , and Gopalan RajaramanChemistry - A European Journal, Jul 2020Since the last decade, the focus in the area of single-molecule magnets (SMMs) has been shifting constructively towards the development of single-ion magnets (SIMs) based on transition metals and lanthanides. Although ground-breaking results have been witnessed for DyIII-based SIMs, significant results have also been obtained for some mononuclear transition metal SIMs. Among others, studies based on CoII ion are very prominent as they often exhibit high magnetic anisotropy or zero-field splitting parameters and offer a large barrier height for magnetisation reversal. Although CoII possibly holds the record for having the largest number of zero-field SIMs known for any transition metal ion, controlling the magnetic anisotropy in these systems are is still a challenge. In addition to the modern spectroscopic techniques, theoretical studies, especially ab initio CASSCF/NEVPT2 approaches, have been used to uncover the electronic structure of various CoII SIMs. In this article, with some selected examples, the aim is to showcase how varying the coordination number from two to eight, and the geometry around the CoII centre alters the magnetic anisotropy. This offers some design principles for the experimentalists to target new generation SIMs based on the CoII ion. Additionally, some important FeII/FeIII and NiII complexes exhibiting large magnetic anisotropy and SIM properties are also discussed.
-
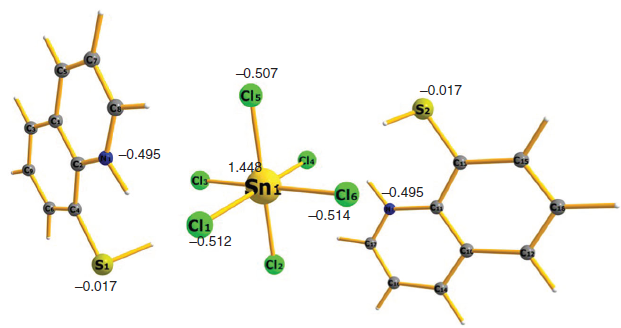 Synthesis, Characterization, and Theoretical Studies of cis -Dichloridobis(8-quinolinethiolato)tin(iv) and bis(8-Sulfanylquinolinium) Hexachloridostannate(iv) DerivativesRajesh Deka , Arup Sarkar, Harkesh B. Singh , Peter C. Junk , David R. Turner , and Glen B. DeaconAustralian Journal of Chemistry, Jul 2020
Synthesis, Characterization, and Theoretical Studies of cis -Dichloridobis(8-quinolinethiolato)tin(iv) and bis(8-Sulfanylquinolinium) Hexachloridostannate(iv) DerivativesRajesh Deka , Arup Sarkar, Harkesh B. Singh , Peter C. Junk , David R. Turner , and Glen B. DeaconAustralian Journal of Chemistry, Jul 2020The structural characterisation of bis(8-sulfanylquinolinium) hexachloridostannate(iv) (2) is reported and the variable reaction behaviour of this compound in different solvents has been explored. In particular, attempted recrystallization of 2 from chloroform and dichloromethane affords two polymorphs of cis-dichloridobis(8-quinolinethiolato)tin(iv), 3m and 3t, respectively. Attempted recrystallization of 2 from methanol gives crystals of 8,8′-dithiodiquinolinium hexachloridostannate(iv) 4. When 2 is dissolved in dimethyl sulfoxide in the presence of air, it undergoes oxidation to afford diquinolinyl-8,8′-disulfide 5. The molecular structures of the isolated compounds 2-4 are unambiguously authenticated by single crystal X-ray diffraction studies. The electronic structure properties of all the isolated compounds 2-4 are thoroughly studied by DFT calculations.
-
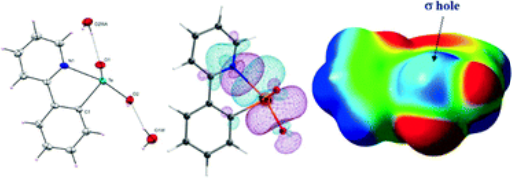
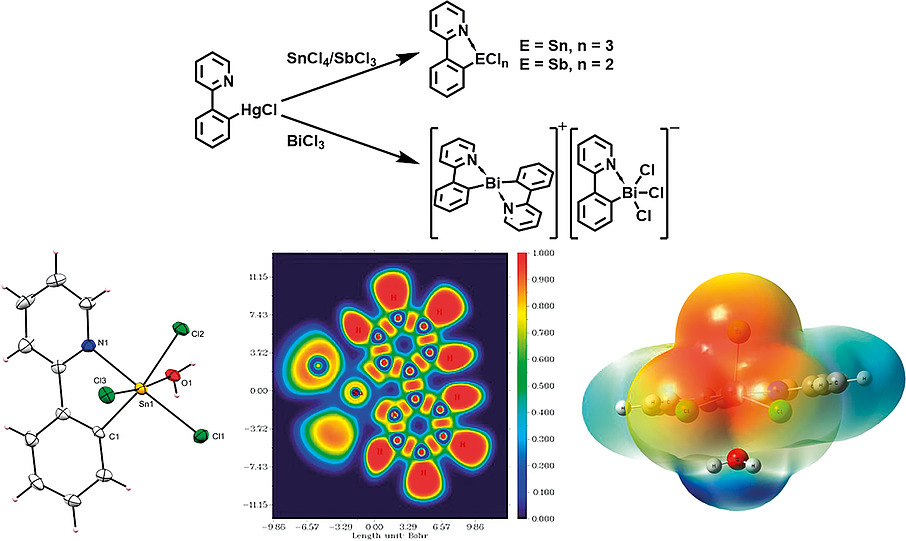 Exploring the Role of Strong Intramolecular Coordination of the 2-(2’-pyridyl)phenyl Group in Heavy Main Group Halides: Insights from Synthesis, Structural, and Bonding AnalysesRajesh Deka , Arup Sarkar, Anand Gupta , Ray J. Butcher , Peter C. Junk , David R. Turner , and 2 more authorsEuropean Journal of Inorganic Chemistry, Apr 2020
Exploring the Role of Strong Intramolecular Coordination of the 2-(2’-pyridyl)phenyl Group in Heavy Main Group Halides: Insights from Synthesis, Structural, and Bonding AnalysesRajesh Deka , Arup Sarkar, Anand Gupta , Ray J. Butcher , Peter C. Junk , David R. Turner , and 2 more authorsEuropean Journal of Inorganic Chemistry, Apr 2020By the transmetallation of ppyHgCl [ppy = 2-(2’-pyridyl)phenyl] (1) with SnCl4 and SbCl3, the synthesis of the monoorgano main group halides, [ppySnCl3(H2O)]\textperiodcentered2(0.5diox) (diox = 1,4-dioxane) [2(H2O)]\textperiodcentered2(0.5diox) and [ppySbCl2] (3) was achieved. The reaction of 1 with BiCl3 does not give the analogous monoorganobismuth dichloride. Instead, it results in the formation of [(ppy)2Bi]+\textperiodcentered[ppyBiCl3]– (4). Reactions of (ppy)2Te and (ppyTe)2 with SO2Cl2 and Br2 result in the formation of ppyTeCl (5) and ppyTeBr (6) respectively. All the synthesized compounds were characterized by multi-nuclear NMR spectroscopy (1H, 13C, 119Sn, and 125Te), FT-IR spectroscopy, ESI-MS, CHN analysis, and single-crystal X-ray diffraction studies. The molecular structures of 2–6 reveal strong intramolecular coordination of the N-atoms of the pendant arms with the main group element centers. DFT calculations; including Natural Bond Order (NBO), Electron Localization Function (ELF), Electrostatic Surface Potential (ESP) and Atoms in Molecules (AIM) infer that the stability of 2–6 relies on the σ-hole participation of the element center with the N-lone pair of the pyridyl arm.
-
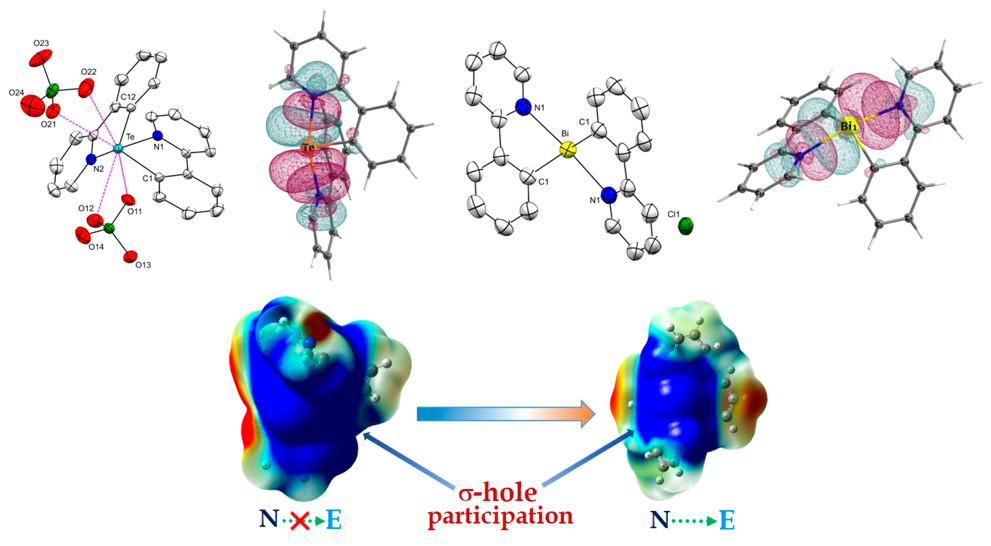 Isolation of Homoleptic Dicationic Tellurium and Monocationic Bismuth Analogues of Non-N-Heterocyclic Carbene DerivativesRajesh Deka , Arup Sarkar, Ray J. Butcher , Peter C. Junk , David R. Turner , Glen B. Deacon , and 1 more authorOrganometallics, Jan 2020
Isolation of Homoleptic Dicationic Tellurium and Monocationic Bismuth Analogues of Non-N-Heterocyclic Carbene DerivativesRajesh Deka , Arup Sarkar, Ray J. Butcher , Peter C. Junk , David R. Turner , Glen B. Deacon , and 1 more authorOrganometallics, Jan 2020The first examples of Te analogues of non-N-heterocyclic carbene (non-NHC) derivatives, [(ppy)2Te]\textperiodcentered2ClO4, [4]\textperiodcentered2ClO4, and[(ppy)2Te]\textperiodcentered2OTf, [5]\textperiodcentered2OTf [where ppy = 2-(2′-pyridyl)phenyl and Tf = O2SCF3] are reported by the metathesis reaction of diorganoiodotelluronium(IV) cation, [(ppy)2TeI]\textperiodcenteredI3, [3]\textperiodcenteredI3, with AgClO4 and AgOTf, respectively. The metathesis reaction of ppyTeCl3, 6, with an excess of AgClO4 resulted in the isolation of [ppyTe(μ-O)]2\textperiodcentered2ClO4, [8]\textperiodcentered2ClO4. The reaction of triorganotelluronium(IV) cation [(ppy)3Te]\textperiodcenteredBr, [10]Br, with K2PdCl4 afforded [(ppy)2TeCl]\textperiodcentered[(ppy)PdCl2], 11. The generality of the "ppy" group on stabilizing other main-group non-NHC analogues has been further established by synthesizing the second example of a Bi analogue of a non-NHC derivative, namely, bismuthenium ion, [(ppy)2Bi]\textperiodcenteredCl, [12]\textperiodcenteredCl, using the same aryl group. All of the synthesized compounds are unambiguously authenticated by single-crystal X-ray diffraction studies. DFT calculations [natural bond orbital (NBO), atoms in molecules (AIM), and electron localization function (ELF)] indicate that the stability of the non-NHC carbenoids relies on the σ-hole participation of the Te/Bi atom with the strong intramolecular coordination ability of the pyridyl N atom of the aryl substituent.
-
 Modulating magnetic anisotropy in Ln(iii) single-ion magnets using an external electric fieldArup Sarkar, and Gopalan RajaramanChemical Science, Aug 2020
Modulating magnetic anisotropy in Ln(iii) single-ion magnets using an external electric fieldArup Sarkar, and Gopalan RajaramanChemical Science, Aug 2020Single-molecule magnets have potential uses in several nanotechnology applications, including high-density information storage devices, the realisation of which lies in enhancing the barrier height for magnetisation reversal (Ueff). However, Ln(iii) single-ion magnets (SIMs) that have been reported recently reveal that the maximum value ofUeffvalues that can be obtained by modulating the ligand fields has already been achieved. Here, we have explored, using a combination of DFT andab initioCASSCF calculations, a unique way to enhance the magnetisation reversal barrier using an oriented external electric field in three well-known Ln(iii) single-ion magnets: [Dy(Py)5(OtBu)2]+(1), [ErN(SiMe3)23Cl]−(2) and [Dy(CpMe3)Cl] (3). Our study reveals that, for apt molecules, if the appropriate direction and values of the electric fields are chosen, the barrier height can be enhanced by twice that of the limit set by the ligand field. The application of an electric field along the equatorial direction was found to be suitable for oblate shaped Dy(iii) complexes and an electric field along the axial direction was found to enhance the barrier height for a prolate Er(iii) complex. For complexes2and3, the external electric field was able to magnify the barrier height to 2-3 times that of the original complexes. However, a moderate enhancement was noticed after application of the external electric field in the case of complex1. This novel non-chemical fine-tuning approach to modulate magnetic anisotropy is expected to yield a new generation of SIMs.
-
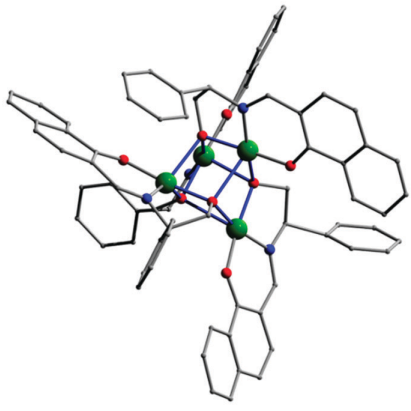 Chiral tetranuclear copper(ii) complexes: Synthesis, optical and magnetic propertiesNaushad Ahmed , Shalini Tripathi , Arup Sarkar, Kamal Uddin Ansari , Chinmoy Das , Neetu Prajesh , and 3 more authorsNew Journal of Chemistry, Aug 2020
Chiral tetranuclear copper(ii) complexes: Synthesis, optical and magnetic propertiesNaushad Ahmed , Shalini Tripathi , Arup Sarkar, Kamal Uddin Ansari , Chinmoy Das , Neetu Prajesh , and 3 more authorsNew Journal of Chemistry, Aug 2020Treating the enantiomerically pure Schiff base ligand (R-H2L1 or S-H2L1) with equimolar copper nitrate led to the isolation of two chiral tetranuclear Cu(ii) cubane complexes with the general molecular formula [Cu4(R-L1)4] (R-1) and [Cu4(S-L1)4] (S-1), which are characterized by single-crystal X-ray diffraction. Both R-1 and S-1 are structurally analogous to each other, which is reflected from their unit cell parameters. The chiral ligands employed efficiently transfer the chirality to the metal complex, which is reflected from their flack parameters and the complex R-1 crystallizes in the chiral, polar point/space group (C2). The chirality is maintained not only in the solid-state but also in the solution state for both R/S-H2L1 and R/S-1. The direct current magnetic susceptibility measurements performed on a polycrystalline sample of a representative complex R-1 exhibits a dominant ferromagnetic interaction resulting in an S = 2 ground state, which is in contrast to the other alkoxide bridged [Cu4O4] complexes reported. The electronic structure and the observed ferromagnetic exchange coupling in R-1 are rationalized by detailed theoretical calculations. This journal is
-
 A large axial magnetic anisotropy in trigonal bipyramidal Fe(ii)Moya A. Hay , Arup Sarkar, Gavin A. Craig , Katie E.R. Marriott , Claire Wilson , Gopalan Rajaraman , and 1 more authorChemical Communications, May 2020
A large axial magnetic anisotropy in trigonal bipyramidal Fe(ii)Moya A. Hay , Arup Sarkar, Gavin A. Craig , Katie E.R. Marriott , Claire Wilson , Gopalan Rajaraman , and 1 more authorChemical Communications, May 2020The first trigonal bipyramidal Fe(ii) complex to display slow relaxation of magnetisation has been isolated, with this behaviour found to arise through a combination of a large magnetic anisotropy (D =-27.5 cm-1) and a pseudo-D3h symmetry at the Fe(ii) centre, as investigated through ab initio and magnetic studies.
-
 Influence of ligand field on magnetic anisotropy in a family of pentacoordinate CoII complexesJoydev Acharya , Arup Sarkar, Pawan Kumar , Vierandra Kumar , Jessica Flores Gonzalez , Olivier Cador , and 3 more authorsDalton Transactions, Mar 2020
Influence of ligand field on magnetic anisotropy in a family of pentacoordinate CoII complexesJoydev Acharya , Arup Sarkar, Pawan Kumar , Vierandra Kumar , Jessica Flores Gonzalez , Olivier Cador , and 3 more authorsDalton Transactions, Mar 2020A family of mononuclear penta-coordinated CoII complexes, [Co(L)Cl2]\textperiodcenteredCH3OH (1), [Co(L)Br2] (2) and [Co(L)(NCS)2] (3) (where L is 1-mesityl-N,N-bis(pyridin-2-ylmethyl)methanamine) were synthesized and characterized. In these complexes, the neutral non-planar ligand, L, binds to three coordination sites around the metal center while two others are bound by anionic halide/pseudo halide ligands. The coordination geometry of the complexes is dictated by the coordinated anionic ligands. Thus, the coordination geometry around the metal ion is distorted trigonal bipyramidal for complexes 1 and 3, while it is distorted square pyramidal for complex 2. Ab initio CASSCF/NEVPT2 calculations on the complexes reveal the presence of an easy plane magnetic anisotropy with the D and E/D values being, 13.3 and 0.14 cm-1 for 1; 36.1 and 0.24 cm-1 for 2 and ±8.6 and 0.32 cm-1 for 3. These values are in good agreement with the values that were extracted from the experimental DC data. AC magnetic measurements reveal the presence of a field-induced slow relaxation of magnetization. However, clear maxima in the out-of-phase susceptibility curves were not observed for 1 and 3. For complex 2, peak maxima were observed when the measurements were carried out under an applied field of 1400 Oe which allowed an analysis of the dynamics of the slow relaxation of magnetization. This revealed that the relaxation is mainly controlled by the Raman and direct processes with the values of the parameters found to be: B = 0.77(15) s-1 K-6.35, n = 6.35(12) and A = 3.41(4) × 10-10 s-1 Oe-4 K-1 and m = 4 (fixed). The ab initio calculation which showed the multifunctional nature of the electronic states of the complexes justifies the absence of zero-field SIM behaviour of the complexes. The magnitude and sign of the D and E values and their relationship with the covalency of the metal-ligand bonds was analysed by the CASSCF/NEVPT2 as well as AILFT calculations.
-
 Isolation of the novel example of a monomeric organotellurinic acidRajesh Deka , Arup Sarkar, Ray J. Butcher , Peter C. Junk , David R. Turner , Glen B. Deacon , and 1 more authorDalton Transactions, Jan 2020
Isolation of the novel example of a monomeric organotellurinic acidRajesh Deka , Arup Sarkar, Ray J. Butcher , Peter C. Junk , David R. Turner , Glen B. Deacon , and 1 more authorDalton Transactions, Jan 2020The stoichiometrically controlled alkaline hydrolysis of 4, (ppy)TeCl3, [ppy = 2-(2′-pyridyl)phenyl] afforded the partially hydrolyzed μ-oxo-bridged dinuclear telluroxane 5, [(ppyTeCl2)2(μ-O)] and a novel example of an Intramolecular Chalcogen Bonding (IChB) stabilized, monomeric organotellurinic acid 6, (ppy)Te(O)OH. The oxidation of diaryl ditelluride 7, (ppyTe)2 using H2O2 resulted in the isolation of μ-oxo-bridged dimethyl ester 8, [(ppy)Te(O)(OH)(OMe)]2(O). The molecular structures of 4-6 and 8 are unambiguously authenticated by single crystal X-ray diffraction studies. The electronic structure of monomeric tellurinic acid 6 is investigated using DFT calculations. The Natural Bond Order (NBO) analysis, in corroboration with Atoms in Molecules (AIM) analysis reveals that tellurinic acid 6 is stabilized by σ-hole participation of the tellurium atom with the pyridyl N-atom resulting in strong electrostatic interactions between the N and Te atoms.










2019
-
 Magnetic Anisotropy in CoIIX4 (X=O, S, Se) Single-Ion Magnets: Role of Structural Distortions versus Heavy Atom EffectArup Sarkar, Subrata Tewary , Shwetali Sinkar , and Gopalan RajaramanChemistry - An Asian Journal, Sep 2019
Magnetic Anisotropy in CoIIX4 (X=O, S, Se) Single-Ion Magnets: Role of Structural Distortions versus Heavy Atom EffectArup Sarkar, Subrata Tewary , Shwetali Sinkar , and Gopalan RajaramanChemistry - An Asian Journal, Sep 2019Mononuclear four coordinate CoII complexes have drawn a great deal of attention as they often exhibit excellent single-ion magnet (SIM) properties. Among the reported complexes, the axial zero-field splitting parameter (D) was found to vary drastically both in terms of the sign as well as strength. There are various proposals in this respect such as structural distortions, heavier atom substitution, metal-ligand covalency, tuning secondary coordination sphere, etc. that are expected to control the D values. To assess the importance of structural distortions vs. heavier atom substitution effect, here we have undertaken detailed theoretical studies based on the ab initio CASSCF/NEVPT2 method to estimate zero-field splitting parameters for twelve complexes reported in the literature. Our test set includes the CoIIX4 (where X=O, S, Se) core structure where the D value was found to vary from +19 to −118 cm−1. Based on the structural variation, we have classified the complexes into three types (I–III) where type I complexes were found to exhibit the largest negative D value as desired for SIMs. The other two types (II and III) of complexes have been found to be inferior with respect to type I. The secondary coordination sphere was also found to influence D, as substitution on the secondary coordination sphere atom was found to significantly alter the magnitude of D values. Particularly, two structural parameters, namely, the dihedral angle between the two ligand planes and the ∠X-Co-X polar angle were found to heavily influence the sign and strength of D values. Our analysis clearly reveals that these structural factors are much more important than the heavier atom substitution, or metal-ligand covalency. A large variation in the D and E/D values among these complexes despite possessing a very close structural similarity offers an exquisite playground for a chemist to design and develop new-generation CoII-based SIMs.
-
 Role of Ab Initio Calculations in the Design and Development of Organometallic Lanthanide-Based Single-Molecule MagnetsAbinash Swain , Arup Sarkar, and Gopalan RajaramanChemistry - An Asian Journal, Sep 2019
Role of Ab Initio Calculations in the Design and Development of Organometallic Lanthanide-Based Single-Molecule MagnetsAbinash Swain , Arup Sarkar, and Gopalan RajaramanChemistry - An Asian Journal, Sep 2019Single-molecule magnets based on lanthanides are very attractive due to their potential applications proposed in the area of microelectronic devices. Very recent advances in this area are due to the blend of conventional lanthanide chemistry with organometallic ligands, and several breakthrough achievements are attained with this combination. Ab initio methods based on multi-reference CASSCF calculations are playing a vital role in the design and development of such molecules. In this minireview, we aim to appraise various contributions in the area of organometallic lanthanide complexes (those containing lanthanide-carbon bonds) and describe how these robust wavefunction-based methods have played a constructive role not only in rationalizing the observed magnetic properties but also proven to be a potential predictive tool with some selected examples.
-
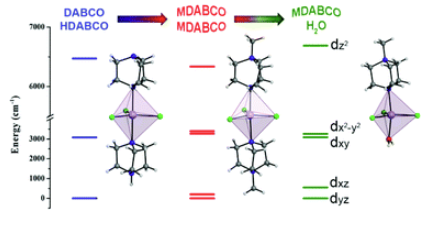 Investigation of the magnetic anisotropy in a series of trigonal bipyramidal Mn(ii) complexesMoya A. Hay , Arup Sarkar, Katie E.R. Marriott , Claire Wilson , Gopalan Rajaraman , and Mark MurrieDalton Transactions, Jun 2019
Investigation of the magnetic anisotropy in a series of trigonal bipyramidal Mn(ii) complexesMoya A. Hay , Arup Sarkar, Katie E.R. Marriott , Claire Wilson , Gopalan Rajaraman , and Mark MurrieDalton Transactions, Jun 2019Understanding how the magnetic anisotropy in simple coordination complexes can be manipulated is instrumental to the development of single-molecule magnets (SMMs). Clear strategies can then be designed to control both the axial and transverse contributions to the magnetic anisotropy in such compounds, and allow them to reach their full potential. Here we show a strategy for boosting the magnetic anisotropy in a series of trigonal bipyramidal Mn(ii) complexes-[MnCl3(HDABCO)(DABCO)] (1), [MnCl3(MDABCO)2]\textperiodcentered[ClO4] (2), and [MnCl3(H2O)(MDABCO)] (3). These have been successfully synthesised using the monodentate [DABCO] and [MDABCO]+ ligands. Through static (DC) magnetic measurements and detailed theoretical investigation using ab initio methods, the magnetic anisotropy of each system has been studied. The calculations reveal that the rhombic zero-field splitting (ZFS) term (E) can be tuned as the symmetry around the Mn(ii) ion is changed. Furthermore, an in silico investigation reveals a strategy to increase the axial ZFS parameter (D) of trigonal bipyramidal Mn(ii) by an order of magnitude.
-
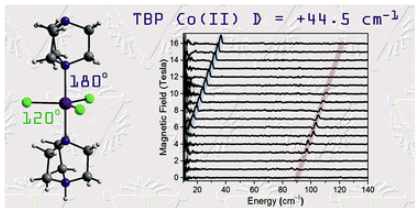 In-depth investigation of large axial magnetic anisotropy in monometallic 3d complexes using frequency domain magnetic resonance and: Ab initio methods: A study of trigonal bipyramidal Co(ii)Moya A. Hay , Arup Sarkar, Gavin A. Craig , Lakshmi Bhaskaran , Joscha Nehrkorn , Mykhailo Ozerov , and 5 more authorsChemical Science, May 2019
In-depth investigation of large axial magnetic anisotropy in monometallic 3d complexes using frequency domain magnetic resonance and: Ab initio methods: A study of trigonal bipyramidal Co(ii)Moya A. Hay , Arup Sarkar, Gavin A. Craig , Lakshmi Bhaskaran , Joscha Nehrkorn , Mykhailo Ozerov , and 5 more authorsChemical Science, May 2019The magnetic properties of 3d monometallic complexes can be tuned through geometric control, owing to their synthetic accessibility and relative structural simplicity. Monodentate ligands offer great potential for fine-tuning the coordination environment to engineer both the axial and rhombic zero-field splitting (ZFS) parameters. In [CoCl3(DABCO)(HDABCO)] (1), the trigonal bipyramidal Co(ii) centre has two bulky axial ligands and three equatorial chloride ligands. An in-depth experimental and theoretical study of 1 reveals a large easy-plane magnetic anisotropy (+ve D) with a negligible rhombic zero-field splitting (E) due to the strict axial symmetry imposed by the C3 symmetric ligand and trigonal space group. The large easy-plane magnetic anisotropy (D = +44.5 cm-1) is directly deduced using high-field EPR and frequency-domain magnetic resonance (FDMR) studies. Ab initio calculations reveal a large positive contribution to the D term arising from ground state/excited state mixing of the 4E′′ states at ∼4085 cm-1 and a minor contribution from the spin-flip transition as well. The nature of the slow relaxation in 1 is elucidated through analysis of the rates of relaxation of magnetisation, taking into account Raman and direct spin-lattice relaxation processes and Quantum Tunnelling of the Magnetisation (QTM). The terms relating to the direct process and QTM were found based on the fit of the field-dependence of τat 2 K. Subsequently, these were used as fixed parameters in the fit of the temperature-dependence of τto obtain the Raman terms. This experimental-theoretical investigation provides further insight into the power of FDMR and ab initio methods for the thorough investigation of magnetic anisotropy. Thus, these results contribute to design criteria for high magnetic anisotropy systems.
-
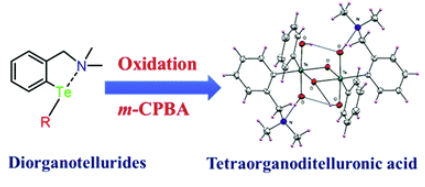 Oxidation behavior of intramolecularly coordinated unsymmetrical diorganotellurides: Isolation of novel tetraorganoditelluronic acids, [RR′Te(μ-O)(OH)2]2Anand Gupta , Rajesh Deka , Arup Sarkar, Harkesh B. Singh , and Ray J. ButcherDalton Transactions, Jun 2019
Oxidation behavior of intramolecularly coordinated unsymmetrical diorganotellurides: Isolation of novel tetraorganoditelluronic acids, [RR′Te(μ-O)(OH)2]2Anand Gupta , Rajesh Deka , Arup Sarkar, Harkesh B. Singh , and Ray J. ButcherDalton Transactions, Jun 2019The oxidation reaction of unsymmetrical diorganotellurides, namely, bis[2-(dimethylamino)methylaryl]tellurides [aryl = phenyl (6), 2-methylphenyl (7), 2,6-dimethylphenyl (8) and 2,6-diisopropylphenyl (9)] with meta-chloroperbenzoic acid afforded the first examples of tetraorganoditelluronic acids, [RR′Te(μ-O)(OH)2]2, where R = 2-NMe2CH2C6H4, R′ = C6H5 (10), 2-MeC6H4 (11), 2,6-MeC6H3 (12) and 2,6-iPrC6H3 (13). The structures of tetraorganoditelluronic acids 10-13 were authenticated by single crystal X-ray diffraction studies. From the molecular structures of 10-13, it was observed that the sp3 N-donor atoms, which were initially involved in intramolecular Te⋯N bonding interactions in diorganotellurides 6-9, did not interact with the tellurium atoms in tetraorganoditelluronic acids 10-13. The 125Te chemical shifts for 10-13 were considerably downfield shifted as compared with the values observed for the corresponding tellurides 6-9. The relative stabilities of the tetraorganoditelluronic acids 10-13 with respect to their lighter analogues (S and Se) have been assessed using DFT calculations.





2018
-
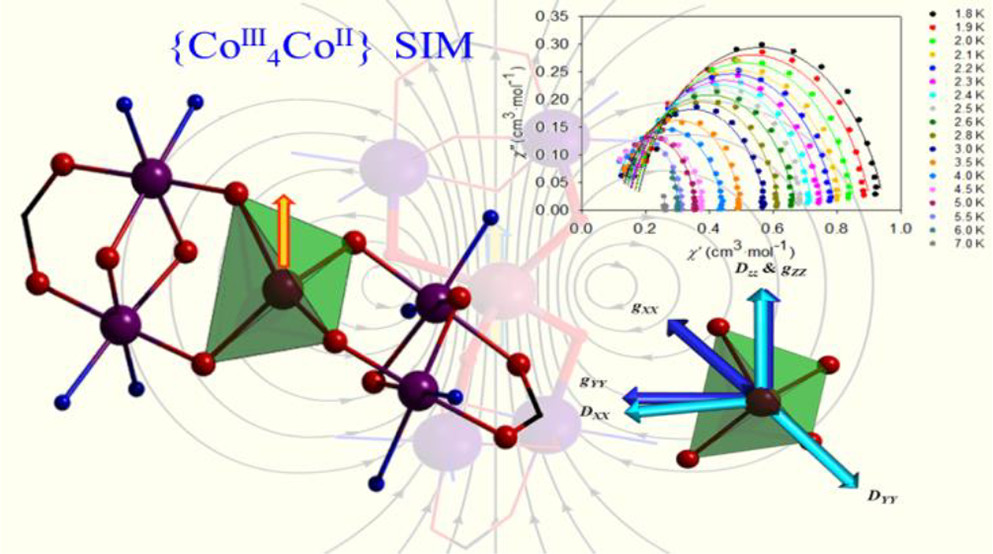 Trapping of a Pseudotetrahedral CoIIO4 Core in Mixed-Valence Mixed-Geometry [Co5] Coordination Aggregates: Synthetic Marvel, Structures, and MagnetismKrishna Chattopadhyay , María José Heras Ojea , Arup Sarkar, Mark Murrie , Gopalan Rajaraman , and Debashis RayInorganic Chemistry, Oct 2018
Trapping of a Pseudotetrahedral CoIIO4 Core in Mixed-Valence Mixed-Geometry [Co5] Coordination Aggregates: Synthetic Marvel, Structures, and MagnetismKrishna Chattopadhyay , María José Heras Ojea , Arup Sarkar, Mark Murrie , Gopalan Rajaraman , and Debashis RayInorganic Chemistry, Oct 2018A systematic one-step one-pot multicomponent reaction of Co(ClO4)2\textperiodcentered6H2O, H3L (2,6-bis((2-(2-hydroxyethylamino)ethylimino)methyl)-4-methylphenol), and readily available carboxylate salts (RCO2Na; R = CH3, C2H5) resulted in the two structurally novel coordination aggregates [CoIICoIII4L2(\mu1,3-O2CCH3)2(μ-OH)2](ClO4)4\textperiodcentered4H2O (1) and [CoIICoIII4L2(\mu1,3-O2CC2H5)2(μ-OH)(μ-OMe)](ClO4)4\textperiodcentered5H2O (2). At room temperature, reactions of H3L in MeOH with cobalt(II) perchlorate salts led to coassembly of initially formed ligand-bound CoII2 fragments following aerial oxidation of metal centers and bridging by in situ generated hydroxido/alkoxido groups and added carboxylate anions. Available alkoxido arms of the initially formed L(\mu1,3-O2CCH3)(μ-OH/OMe)Co2+ fragments were utilized to trap a central CoII ion during the formation of [Co5] aggregates. In the solid state, both complexes have been characterized by X-ray crystallography, variable-temperature magnetic measurements, and theoretical studies. Both 1 and 2 show field-induced slow magnetic relaxation that arises from the single pseudo-Td CoII ion present. The structural distortion leads to an easy-axis magnetic anisotropy (D = -31.31 cm-1 for 1 and -21.88 cm-1 for 2) and a small but non-negligible transverse component (E/D = 0.11 for 1 and 0.08 for 2). The theoretical studies also reveal how the O-Co-O bond angles and the interplanar angles control D and E values in 1 and 2. The presence of two diamagnetic Co2(μ-L) hosts controls the distortion of the central CoO4 unit, highlighting a strategy to control single-ion magnetic anisotropy by trapping single ions within a diamagnetic coordination environment.
-
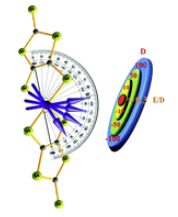 Deciphering the origin of invariance in magnetic anisotropy in [FeIIS4] complexes: a theoretical perspectiveArup Sarkar, Gunasekaran Velmurugan , Thayalan Rajeshkumar , and Gopalan RajaramanDalton Transactions, Jun 2018
Deciphering the origin of invariance in magnetic anisotropy in [FeIIS4] complexes: a theoretical perspectiveArup Sarkar, Gunasekaran Velmurugan , Thayalan Rajeshkumar , and Gopalan RajaramanDalton Transactions, Jun 2018For q-bit applications based on coordination complexes, invariance in zero-field splitting parameters upon structural distortions is desired. Here, by employing ab initio calculations, we have probed the origin of such resistance observed in four coordinate [FeII(C3S5)2]2- complexes. While unaltered D parameters are noted for a short range of structural distortions such as dihedral angle, if a wider range is chosen, larger variations are prominent in both D and E/D values.
-
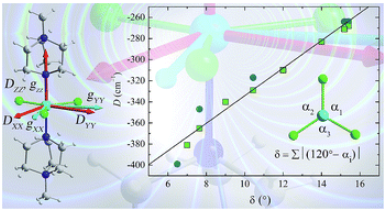 Probing the origin of the giant magnetic anisotropy in trigonal bipyramidal Ni(ii) under high pressureGavin A. Craig , Arup Sarkar, Christopher H. Woodall , Moya A. Hay , Katie E.R. Marriott , Konstantin V. Kamenev , and 5 more authorsChemical Science, Jan 2018
Probing the origin of the giant magnetic anisotropy in trigonal bipyramidal Ni(ii) under high pressureGavin A. Craig , Arup Sarkar, Christopher H. Woodall , Moya A. Hay , Katie E.R. Marriott , Konstantin V. Kamenev , and 5 more authorsChemical Science, Jan 2018Understanding and controlling magnetic anisotropy at the level of a single metal ion is vital if the miniaturisation of data storage is to continue to evolve into transformative technologies. Magnetic anisotropy is essential for a molecule-based magnetic memory as it pins the magnetic moment of a metal ion along the easy axis. Devices will require deposition of magnetic molecules on surfaces, where changes in molecular structure can significantly alter magnetic properties. Furthermore, if we are to use coordination complexes with high magnetic anisotropy as building blocks for larger systems we need to know how magnetic anisotropy is affected by structural distortions. Here we study a trigonal bipyramidal nickel(ii) complex where a giant magnetic anisotropy of several hundred wavenumbers can be engineered. By using high pressure, we show how the magnetic anisotropy is strongly influenced by small structural distortions. Using a combination of high pressure X-ray diffraction, ab initio methods and high pressure magnetic measurements, we find that hydrostatic pressure lowers both the trigonal symmetry and axial anisotropy, while increasing the rhombic anisotropy. The ligand-metal-ligand angles in the equatorial plane are found to play a crucial role in tuning the energy separation between the dx2-y2 and dxy orbitals, which is the determining factor that controls the magnitude of the axial anisotropy. These results demonstrate that the combination of high pressure techniques with ab initio studies is a powerful tool that gives a unique insight into the design of systems that show giant magnetic anisotropy.


